Az elektroforézis olyan elválasztási eljárás, melyet kis- és nagy-méretű töltött részecskék, biomolekulák (peptidek, fehérjék, nukleinsavak, szénhidrátok), vírusok és sejtek elválasztására használnak. Előnye más technikákkal szemben a nagy felbontóképesség, az érzékenység, a vizes közeg, a gyors elválasztás, kis anyagmennyiség-igény illetve az, hogy sokszor nem szükséges a mintát előzetesen megtisztítani a szennyeződésektől, ami jelentős költség- és időmegtakarítást jelent.
Az elektroforézis (gör. elektro ‘áram’ + forézis ‘szállít’) alapja, hogy egy keverék oldat töltéssel rendelkező komponensei elektromos erőtérben különböző sebességgel mozognak. Ennek gyakorlati megvalósítására kezdetben különböző hordozókat (szűrőpapír, vékonyréteglap, cellulózacetát-membrán), illetve lágy géleket (keményítő, agar, poliakrilamid) alkalmaztak. A 80-as évektől ellenben kezdett elterjedni az elektrolittal töltött kapilláris csövekben végzett elektroforézis is. A nagy térerő (akár 1000V/cm) alkalmazása miatt rendkívüli szelektivitás és kivételes hatékonyság érhető el, így a kapilláris elektroforézis napjaink egyik legdinamikusabban fejlődő analitikai módszerének tekinthető.
Az elválasztás elve alapján az elektroforetikus módszerek a következőképp csoportosíthatók: kapilláris zónaelektroforézis (CZE), micelláris elektrokinetikus kromatográfia (MEKC), kapilláris gélelektroforézis (CGE), kapilláris izoelektromos fókuszálás (CIEF), kapilláris elektrokromatográfia (CEC) és kapilláris izotachoforézis (CITP).
Az elektroforézis elvén alapuló elválasztási technikák esetén az oldat töltéssel rendelkező részecskéi elektromos erőtér hatására különböző sebességgel mozdulnak el. Az elektroforézis elméleti alapjait Kohlrausch fektette le a XIX. század végén, amikor egyenleteivel leírta az ionok elektrolitban való vándorlását. Az első kísérleteket Tiselius hajtotta végre 1937-ben, amikor egy U alakú csőbe fehérjeoldatot töltött, mely fölé – mindkét szárba – elektrolitoldatot rétegzett (úgy, hogy köztük éles határfelület maradjon), majd elektromos feszültséget kapcsolt a csővégekre. Ekkor azt tapasztalta, hogy a különböző fehérjék töltésüktől és mozgékonyságuktól függően különböző sebességgel, különböző irányokba vándorolnak (a minta vándorlását a határfelület elmozdulása jelezte). Munkájáért később Nobel-díjjal jutalmazták.

A zónaelektroforézist oldatokban először Hjertén írta le 1967-ben. Kísérleteihez egy horizontális elhelyezésű, körülbelül 3 mm belső átmérőjű, tengelye körül forgatott kvarc-csövet használt (a tengely körüli forgatásra a gravitáció hatására bekövetkező ülepedés ellensúlyozása miatt volt szükség). Az ennél kisebb (100-200 μm) belső átmérőjű csövekben végzett elektroforézis kísérletek évekkel később, 1980 körül valósultak meg (a kapillárisokban az ülepedés nem játszik szerepet), kifejlesztésük Jorgenson és Lukács nevéhez fűződik.

A kapilláris zónaelektroforézis (CZE) a legegyszerűbb és leggyakrabban alkalmazott kapilláris elektroforetikus módszer, mely a részecskék eltérő elektroforetikus mozgékonyságán alapul, melyet a részecskék eltérő töltés/méret aránya határoz meg. Az elválasztás az elektrolittal töltött, általában 25-75 µm belső átmérőjű kvarckapillárisban történik, amelybe keskeny sávban (zónában) mintát injektálunk, és az alkalmazott feszültség hatására a kezdeti minta zóna komponensei diszkrét zónákba választódnak szét. Az elváló zónákat leggyakrabban UV-látható fényabszorpciós detektor érzékeli. A nagy elektromos térerő használata rövid mérési időt, valamint nagy elválasztási hatékonyságot és felbontást biztosít.
Az elektroozmotikus áramlás (EOF) a töltéssel rendelkező felület (uncoated – nem bevonatos – kapilláris) mentén, elektromos tér hatására kialakuló folyadékáramlás.
A kvarc kapilláris felületén szilanolcsoportok (SiOH) vannak. Az elektrolit pH-ját 2,5 fölé emelve a szilanolcsoportok gyenge sav lévén disszociálni kezdenek, a kapilláris fala negatív töltésűvé válik (SiO-). A töltésegyensúly fenntartása végett az oldatban lévő kationok a kapilláris fala mentén pozitív töltésű kettősréteget képeznek, melyben a szilárd felülethez legközelebb lévő ionokra a síkelrendeződés, attól távolabb pedig diffúz elrendeződés érvényes. A kettősrétegben található ellenionok a fal potenciáljához nagyon közeli potenciált (zéta potenciált) hoznak létre. Ha feszültséget kapcsolunk a kapilláris végeire, a diffúz réteg kationjai hidrátburkukkal együtt a katód irányába vándorolnak. Mivel a kapilláris átmérője kicsi, ezért ez a teljes oldattömeg együttes áramlását eredményezi. Az EOF nagysága és iránya függ a kapillárisban levő oldat jellegétől (pH, ionerősség). A víz nagy dielektromos állandójának figyelembevételével az EOF mozgékonysága a következő képlettel írható le:
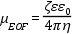
ahol: µEOF = elektoozmotikus
áramlás mozgékonysága ζ = zéta potenciál ε = elektrolit dielektromos
állandója εo = vákuum dielektromos állandója η
= elektrolit viszkozitása
Mivel az elektroozmotikus áramlás hajtóereje egyenletes a kapillárisban, az EOF dugószerű áramlási profilt biztosít (200 µm belső átmérőig!). Ennek köszönhetően az elméleti tányérszám sok esetben meghaladja a 105 értéket. Az elektroozmotikus áramlás ezenkívül valamennyi oldott részecske egyidejű vizsgálatát is biztosítja, függetlenül azok töltésétől.
Az elektroforetikus elválasztási módszerek estében a részecskék vándorlási sebessége (v) arányos az alkalmazott térerővel (E), azaz a hosszegységre eső potenciálváltozással. Az arányossági tényezőt a részecske mozgékonyságának (µe) nevezzük:
v = µe E
v = a részecske sebessége [cm/s]
µe = az elektroforetikus mozgékonyság
[cm2/V·s] E = az elektromos térerő
[V/cm]
Az elektromos erőtérben tehát a különböző töltéssel és mérettel rendelkező részecskék eltérő, de állandó sebességgel vándorolnak, így elkülöníthetők (szeparálhatók) egymástól. A vándorló részecskék sebessége akkor állandó, ha a rájuk ható két, egymással ellentétes erő – Fe elektromos erő és Fs súrlódási erő – egymással egyenlő.
Fe = Fs
qE = 6πηrv
q = a részecske töltése η = az oldat viszkozitása
[Pa·s] r = a részecske sugara [cm]
Az előző egyenletek alapján az elektroforetikus mozgékonyság fizikai állandókkal kifejezve:
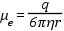
Az egyenletből következik, hogy a kisméretű, nagy töltésű részecskék rendelkeznek a legnagyobb mozgékonysággal. A részecske mobilitása fordítottan arányos a közeg viszkozitásával, de az adott közegben (pH, ionerősség, hőmérséklet, stb.) a mobilitás állandó és az adott részecskére jellemző érték.
A vándorló részecskék elektrontöbblettel bíró negatív töltésű anionok (az anód felé mozognak) vagy elektronhiányos pozitív töltésű kationok (a katód felé mozognak). Az egyes komponensek vándorlási irányának meghatározásához azonban kétféle mozgékonyságot kell figyelembe vennünk: 1) az elektroozmotikus áramlás mozgékonysága (µEOF), 2) az adott komponens saját töltéséből eredő elektroforetikus mozgékonyság (µi). A két mozgékonyság vektori összegzéséből adódik az eredő, ún. látszólagos mozgékonyság (µl):
µl = µi + µEOF
A µEOF egy semleges molekula (pl. aceton) mozgékonyságával egyezik meg, így annak hozzáadásával meghatározható.
Általában az EOF egy nagyságrenddel nagyobb, mint az oldott komponensek vándorlási sebessége (vEOF»µiE), így a kationok, anionok és semleges részecskék is a katód (negatív pólus) felé haladnak, ezáltal egyidejűleg vizsgálhatók.
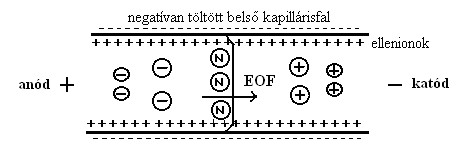
Az anódtól a katód irányába történő részecskeáramláskor tehát legelöl a kationok vándorolnak (az elektroforetikus vonzóerő és az EOF is mozgatja őket), a kationok után vándorló töltés nélküli (neutrális) vegyületeket csak az EOF szállítja (ezek nem szeparálódnak, mivel nincs elektroforetikus mozgékonyságuk), a semleges részecskéket pedig az anionok követik (melyek szintén a negatív pólus felé mozognak, ha az EOF szállítóereje nagyobb, mint az azzal ellentétes irányú, rájuk ható elektroforetikus erő).
Vannak olyan esetek is, amikor a minta részecskéi (pl. fehérjék) hajlamosak a kapilláris falára adszorbeálódni, ami csökkenti az elválasztás hatékonyságát, és bizonyos esetekben nagyon lassan csökkenő aszimmetrikus jeleket kapunk. Ilyenkor szükséges az EOF csökkentése vagy kiküszöbölése. A kiküszöbölés egyik módja, hogy a kapilláris falához kovalens kötéssel bizonyos polimereket (pl. poliakrilamid) kapcsolunk (coated kapilláris). Ez a bevonat megakadályozza a kapilláris falára történő fehérje adszorpciót is. Az EOF-t szabályozhatjuk pl. a puffer pH-jának, koncentrációjának és az elektromos tér nagyságának változtatásával.
8.5. ábra - A kapilláris elektroforézis készülék vázlatos felépítése (NF: nagyfeszültségű tápegység)
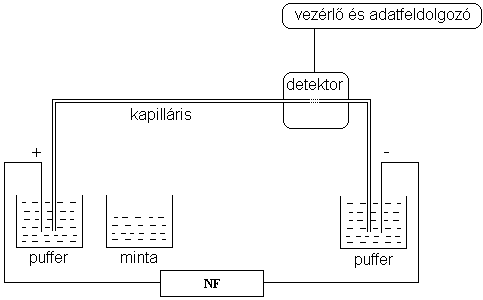
A kvarckapilláris végei két, pufferoldatot tartalmazó edénybe merülnek. Ezen edényekbe nyúlnak az elektródok is, amelyekre nagyfeszültséget kapcsolunk. A minták injektálása a detektortól távolabb lévő kapillárisvégen történik. Az injektálás történhet többek között hidrodinamikusan (nyomás alkalmazásával) és elektrokinetikusan (nagyfeszültségű impulzus hatására bekövetkező elektroforetikus migrációval) is. A minta komponenseinek vándorlása a pufferrel töltött kapillárisban akkor kezdődik, amikor a pufferedényekre feszültséget kapcsolunk. Ez rendszerint 10-30 kV, melynek hatására 5-50 μA áram mérhető. A áramfejlődés közben keletkező Joule-hőt a kis átmérőjű kapilláris nagy felületén keresztül adja le. A detektálás közvetlenül a kapilláris falán keresztül történik. Detektorként pl. UV-látható detektort, lézer indukált fluoreszcens detektort, vezetőképességi detektort, vagy akár tömegspektrométert is alkalmazhatunk. a fényútba eső kapillárisdarabról a védőréteget mérés előtt el kell távolítani
A modern CE készülékek a sorozatmérésekhez rendkívül hasznos automata mintaadagolóval, a kapilláris állandó hőmérsékletét biztosító termosztáló egységgel (a közeg viszkozitása hőmérsékletfüggő!) és a készülék vezérlését, valamint a mérési adatok feldolgozását végző számítógéppel vannak felszerelve.
A kapilláris elektroforézis során elektroferogramot kapunk, ahol a migrációs idő függvényében ábrázolják az abszorbanciát (UV-látható detektor alkalmazásakor). Minőségi információt a csúcsok helye (migrációs idő), illetve az adott anyag mozgékonysága (ami az elválasztás körülményeitől jelentősen függ) szolgáltat. A mennyiségi meghatározásnál a csúcs magasságából vagy területéből következtetünk az anyag koncentrációjára.
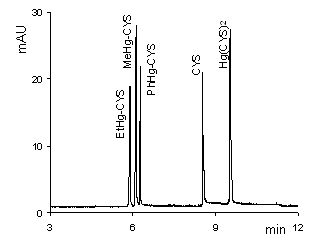

Migrációs idő. A migrációs (vándorlási) idő azt az időtartamot jelöli, mely alatt az oldott anyag eljut a kapilláris elejétől a detektorablakig. Kísérletileg a mozgékonyságot a következő egyenlettel lehet meghatározni:
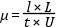
l = a kapilláris effektív hossza (a kapilláris hossza
a detektorig) L = a kapilláris teljes hossza t = migrációs idő U =
alkalmazott feszültség
Elméleti tányérszám (N), tányérmagasság (H). Az elválasztás hatékonyságát jellemzik.
N = 5,54
(t/w1/2)2 H = l/N
t = migrációs idő l = a kapilláris effektív hossza
w1/2 = a kapott csúcs
félértékszélessége
Felbontás (R). Két csúcs relatív elválasztásának a mértéke.
R =
2(t2-t1) /
(w1+w2) w =
csúcsszélesség
A készüléket bekapcsoljuk, üzemképes állapotba hozzuk. A kapilláris tartóba helyezett kapilláris rögzítjük a készülékben. A kapilláris a mérés megkezdése előtt kondícionáltatni (mosatni) kell, először 0,1 M NaOH-oldattal (uncoated kapilláris esetén) és desztillált vízzel, majd a puffer oldattal.
Mérési paraméterek. A gyakorlaton többféle feladat adható, de mindegyiknél elvárás az alábbi paraméterek megadása:
Készülék típusa:
Puffer:
Kapilláris hossza (teljes és effektív):
Injektálás módja, injektált mennyiség:
Hőmérséklet:
Feszültség:
Polaritás:
Detektálás módja, hullámhossza:
Körülmények: Puffer: 20 mM borát, pH=9.4
Injektálás: 100 mbar . s
Hőmérséklet: 25°C
Feszültség: 25 kV
Detektálás: 200 nm, 220 nm, 260 nm
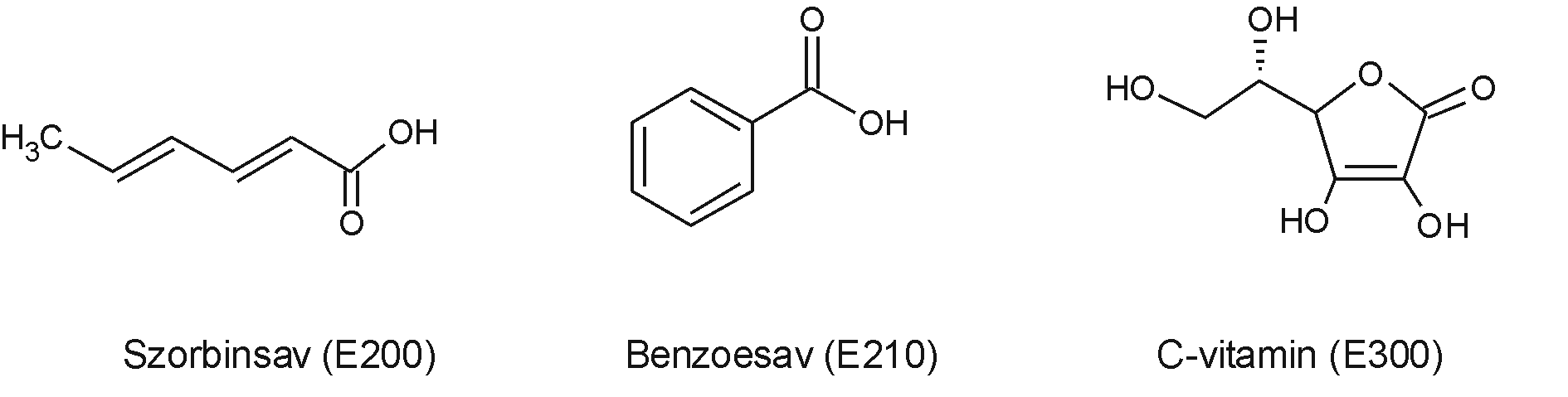
Minták: Ismert koncentrációjú benzoesav, szorbinsav és C-vitamin tartalmú oldat. Citromlé és egyéb ivólevek.
Feladat és beadandó eredmények: Az ismert összetételű minta csúcsainak beazonosítása a töltés/méret arány alapján.
A kapott csúcsok migrációs idejének, a csúcsok félértékszélességének megadása.
Látszólagos mozgékonyságunk kiszámítása.
Az ismert összetételű minta elválasztásának jellemzése tányérszám, tányérmagasság, felbontás megadásával.
A citromlé (vagy más ivólé) tartósítószer (vagy C-vitamin) tartalmának minőségi és mennyiségi meghatározása (a csúcs alatti terület alapján koncentráció meghatározás).
Körülmények: Puffer: 5 mM imidazol, pH=4,5 kénsavval beállítva
Kapilláris: leff= 56 cm, belső átmérő= 50 µm
Injektálás: 100 mbar . s
Hőmérséklet: 25°C
Feszültség: 25 kV
Detektálás: 214 nm, indirekt ("negatív csúcsok")
Minták: 1. K+, Ba2+, Ca2+, Na+, Li+ tartalmú, ismeretlen koncentrációjú oldat.
2. 10, 30, 50 µg/ml Ca2+-tartalmú oldatok.
3. ivóvíz
Feladat és beadandó eredmények: Ca2+ migrációs idejének megadása.
A többi ion migrációs idejének valószínűsítése az elektroferogram és a mozgékonysági adatok alapján (K: 0.68, Na: 0.48, Ca: 0.45, Ba: 0.42 cm2/kVs).
A Ca2+ ismeretlen koncentrációjának meghatározása.
Az ivóvíz fő fémion-tartalmának kvalitatív meghatározása.
Körülmények: Puffer: 10 mM foszfát, pH=2,5 illetve pH=8
Kapilláris: leff= 22 cm, belső átmérő= 50 µm
Injektálás: 2 psi*s
Hőmérséklet: 25°C
Feszültség: 10 kV
Detektálás: 200-300 nm között (5 nm-ként)
Szerves vegyületek törzsoldatai: 1 mg/ml triptofán-metilészter (TME); 1 mg/ml triptofán-etilészter (TEE); 1 mg/ml triptofán-butilészter (TBE)
Minták: A szerves vegyületek keveréke a törzsoldatokból 20-szoros hígítással készül (pl.: 2 µl TME + 2 µl TEE + 2 µl TBE + 34 µl desztillált víz).
Feladat és beadandó eredmények: A kapott csúcsok migrációs idejének, csúcsszélességének, csúcs félértékszélességének megadása.
A csúcsok beazonosítása a töltés/méret arány alapján.
Az elválasztások jellemzése tányérszám, tányérmagasság, felbontás megadásával.
áramlási profil, elektroferogram, elektroozmotikus áramlás, elektroforetikus mobilitás, kapilláris elektroforézis, migrációs idő, nagyfeszültség, térerő, CE, CZE, EOF, UV
Mi az elválasztás alapja a kapilláris zónaelektroforézisnél?
Hogyan adható meg az ion sebessége, és az elektroforetikus mozgékonyság?
Mit nevezünk elektroozmotikus áramlásnak, mik ezen áramlás előnyei (pl. milyen az áramlás profilja)?
Hogyan szabályozható illetve küszöbölhető ki az elektroozmotikus áramlás?
Milyen lesz a részecskék vándorlási sorrendje abban az uncoated kapillárisban, ahol az anódtól a katód irányába történik a részecskeáramlás (az EOF katód irányú)?
Ismertesse a kapilláris elektroforézis készülék felépítését!
Milyen módszerekkel történhet a minta injektálása a kapillárisba?
Milyen detektorokat alkalmazhatunk kapilláris zónaelektroforézisnél?
Hogyan számolunk látszólagos mozgékonyságot, tányérszámot, tányérmagasságot és felbontást?
Az elektroferogramon kapott csúcsok milyen adataiból következtetünk a minőségi és a mennyiségi információkra?