Tartalom
- Bevezetés
- I. Sztereokémia
- II. Szénhidrogének
- 7. Telített szénhidrogének
- 8. Telítetlen szénhidrogének: Alkének
- 9. Telítetlen szénhidrogének: Alkinok
- 10. Aromás szénhidrogének
- III. Halogénezett szénhidrogének és elemorganikus vegyületek
- IV. Alkoholok, fenolok, éterek
- V. Kéntartalmú vegyületek
- 14.
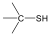
- Tiolok, diszulfidok
- Előfordulásuk
- Tiolok nevezéktana
- Tiolok előállítása
- Tiolok fizikai tulajdonságai
- Tiolok kémiai tulajdonságai, reakciói
- Biológiailag fontos tiolok
- Szulfidok (tioéterek)
- Előfordulásuk
- Nevezéktan
- Elektronszerkezet
- Szulfidok előállítása
- Szulfidok fizikai tulajdonságai
- Szulfidok kémiai reakciói
- Szulfidok biológiailag fontos származékai
- Kérdések, feladatok
- VI. Nitrogéntartalmú vegyületek
- VII. Aldehidek, ketonok
- VIII. Szénhidrátok
- IX. Karbonsavak
- 23. Karbonsavak
- 24. A karbonsavak és karbonsavszármazékok elektronszerkezete:
- 25. Karbonsavak csoportosítása:
- 26. A karbonsavak és karbonsavszármazékok elnevezése:
- 27. Karbonsavak és karbonsavszármazékok előállítása
- 28. A karbonsavak, karbonsavszármazékok fizikai tulajdonságai
- 29. A karbonsavak és karbonsavszármazékok kémiai tulajdonságai
- 30. A karbonsavak és karbonsavszármazékok fontosabb képviselői:
- 31. Szénsavszármazékok
- X. Aminosavak, peptidek, fehérjék
- 32. Aminosavak, peptidek, fehérjék csoportosítása
- 33. α-Aminosavak fizikai tulajdonságai és sav-bázis jellege
- 34. Az α-aminosavak térszerkezete
- 35. Az α-aminosavak előállítása
- 36. Az α-aminosavak reakciói
- 37. Peptidek és fehérjék szerkezete
- 38. A fehérjék csoportosítása
- 39. Peptidek szintézise
- 40. Feladatok
- XI. Öttagú heterociklus vegyületek
- 41. Öttagú heterociklus vegyületek
- 42. Csoportosítás:
- 43. Nevezéktan:
- 44. Öttagú, egy heteroatomot tartalmazó heterociklusok
- 45. Öttagú, több heteroatomot tartalmazó vegyületek:
- 46. Hattagú, egy heteroatomot tartalmazó vegyületek
- 47. Nukleotidok, nukleinsavak
- 48. Feladatok a heterociklusok és nukleinsavak köréből:
- XII. Vitaminok
- XIII. Szerves vegyületek lebomlása, hatása a biológiai környezetre
Az anyagi világról szerzett természettudományos ismerek bővülése magával hozta az egyes tudományok (fizika, matematika, kémia, geológia, biológia, stb.) tagozódásának megkezdődését, amit nem lehet egy konkrét évszámhoz kötni, de több jelentős felfedezés miatt az 1500-as évek közepe jelölhető meg. A kémia ekkor az ún. jatrokémia (orvosi kémia) időszakát élte. 1500-as évek jatrokémikusainak fontos szerepük volt új (mai besorolás szerint szerves) vegyületek elkülönítésében is. Pl. Valerius Cordus (német) 1540-ben felfedezte a sztrichnint tartalmazó növényi magok mérgező hatását, majd alkohol és kénsav reakciójával étert, Andreas Libavius (német) 1595-ben acetont állított elő. A XVII. századot tartják a tudományos kémia kezdetének, amikor különvált az orvostudománytól. A kémia tudományág később tovább tagozódott és a XIX. század elején kialakult a szerves kémia, majd elkezdődött a lendületes fejlődése, ami mind a mai napig tart.
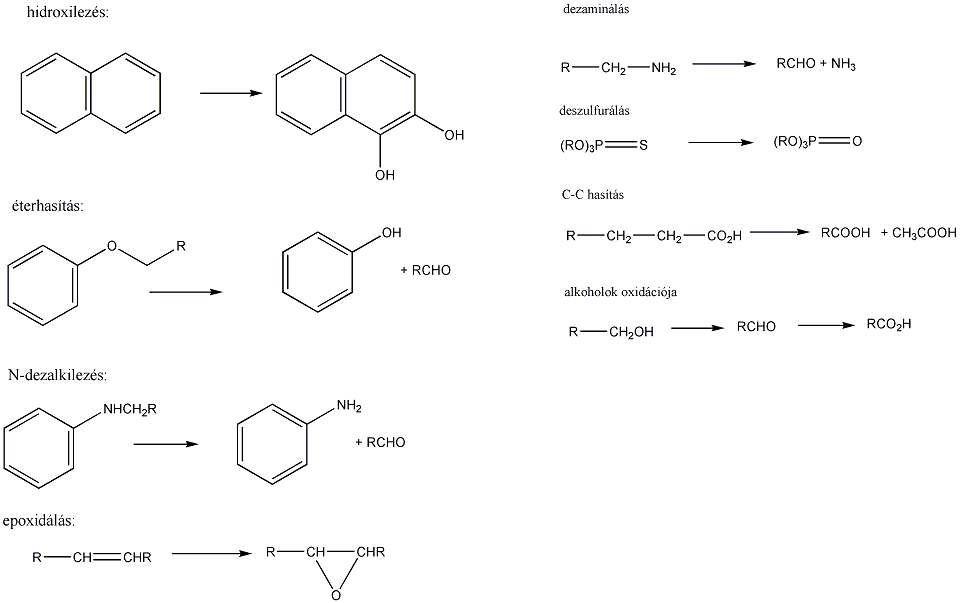
Az 1700-as évek utolsó harmadában a különböző növényi és állati eredetű anyagokat összehasonlítva az ásványi anyagokkal számos hasonlóság mellett jelentős különbségeket is találtak. Kialakult egy olyan rendszerezés, amely az élő szervezetek anyagait organikus vagy „szerves” csoportba sorolta, míg a másik csoportba az anorganikus vagy szervetlen anyagok kerültek. A korabeli felfogás szerint szerves anyagot csak élő szervezet képes létrehozni az ún, vis vitalis = életerő révén, tehát laboratóriumi körülmények között szerves anyag nem állítható elő. Ez a felfogás volt érvényben egészen addig, amíg Wöhler 1824-ben és 1828-ban két híressé vált kísérletet nem végzett: elsőként az anorganikus anyagok közé sorolt Hg(CN)2-t hevítve diciánt nyert, amit hidrolízissel oxálsavvá tudott átalakítani. Az oxálsav korábban ismert volt mint növényi sav, tehát az életerő elmélet szerint laboratóriumi módszerrel nem állítható elő.

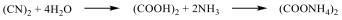
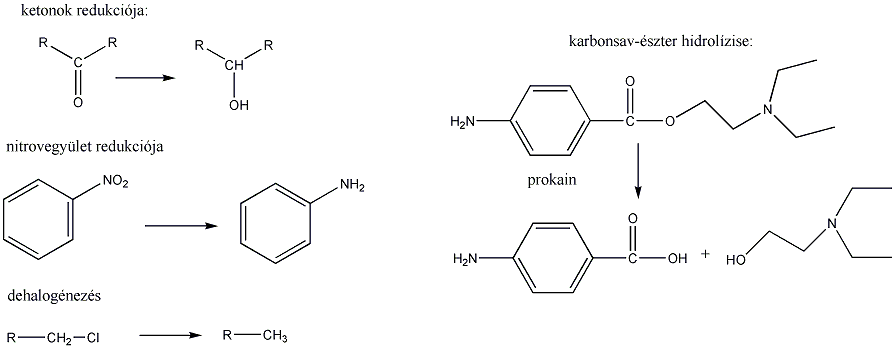
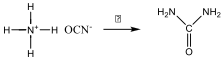
Ugyancsak Wöhler 1828-ban ammónium-cianát hevítésével, korábban már az állati szervezetekből ismert organikus anyagot, karbamidot kapott.
Bár ezekből a kísérletek még nem döntötték meg azonnal a vis vitalis elméletet, de sok hasonló megfigyelés után, főleg Liebig, Bertholet, Dumas, Kolbe, Gerhardt, Wurtz és Kekulé kísérleti munkái nyomán kiderült, hogy nincs elvi különbség az organikus és anorganikus csoportba sorolt anyagok között, mindegyikük előállítható laboratóriumi módszerekkel és érvényesek rájuk az általános kémiai törvényszerűségek. Ennek ellenére célszerű a két csoportot megkülönböztetni, amit alapvetően a szerves vegyületek jóval nagyobb száma, eltérő tulajdonságai indokolnak. A szerves vegyületek többségét nem a természetben előforduló vegyületek képezik, hanem a mesterségesen előállított anyagok adják. Bár a szerves vegyületekben a szénatomok mellett leggyakrabban a H, N, O, S, P és a halogének fordulnak elő, napjainkig szinte valamennyi elem beépítésére sor került. A bő 200 éves múltú tudományos szerves kémia gyors elméleti és gyakorlati fejlődése nélkül, szintetikus módszerei, ipari alkalmazásuk nélkül elképzelhetetlen lenne mindennapi életünk.
A szénvegyületek nagy száma a szénatom tulajdonságaira vezethető vissza, ami egyedülálló lehetőséget jelent, hogy korlátlanul összekapcsolódjanak és kimeríthetetlen számban lehessen újabb és újabb származékokat előállítani. Az elmúlt évtizedekben az ismert szerves vegyületek száma 5-8 évente megduplázódott és már 15 millión felül jár. A szénvegyületek körében az izoméria jelensége az egyik oka a szerves vegyületek nagy számának. Az izoméria miatt az összegképlet általában nem jellemzi kielégítően a szerves vegyületeket, egyértelműen csak szerkezeti képletekkel jellemezhetők.
A szerves vegyületekben az elsődleges kémiai kötések közül – a számos kivétel mellett - leginkább a kovalens kötés jellemző.
A szerves vegyületek szerkezetének megismerésében kiemelkedik Kekulé munkássága, aki felismerte, hogy az addig ismert vegyületekben a szénatom mindig négy "vegyértékű" volt, és a szénatomok egymással is képesek kapcsolódni szénlánc kialakulása közben. Ezt a négy vegyértéket Kekulé még azonos értékűnek tekintette. Ugyancsak Kekulé javasolta a benzol gyűrűs szerkezetét, és a gyűrűben lévő szénatomok egyenértékűségét.
Az atomok egymáshoz kapcsolódására számos ábrázolási módot alkalmaztak. A 1870-es években terjedt el az a ma is használatos írásmód, hogy az atomok közé a vegyértékek számának megfelelő számú kötést vonallal jelölik.
Az 1800-as évek végére kialakultak azok a ma is helytálló alaptételek, amelyek a szerves vegyületek szerkezetelméletének alapjait képezik:
-
a szénatom vegyületeiben mindig négy vegyértékű,
-
ez a négy vegyérték egymással egyenértékű,
-
a szénatom négy vegyértéke a telített vegyületekben tetraéderes térorientációjú,
-
a szénatomok egymással is kapcsolódhatnak, lánc vagy gyűrűs szerkezetű molekulaváz kialakulása közben.
A modern szerkezetelméletének kialakulásában ki kell emelni Le Bel és van't Hoff munkásságát.
A szén a periódusos rendszer 4. oszlopában foglal helyet. Rendszáma 6, tehát az atommagja körül 6 db elektron található. A nemesgázszerkezetet 4 db elektron leadásával, vagy felvételével érhetné el. A tőle balra elhelyezkedő fémek elektropozitív jellege a szén felé haladva csökken, míg a periódusos rendszer jobb oldalán elhelyezkedő elemek (nemfémes elemek) esetében az elektronegatív jelleg csökken a szén irányába haladva. Energetikailag C4+ és C4- ion képződése szinte lehetetlen.
A szénatom emiatt csak úgy tud nemesgáz-konfigurációt elérni vegyületképzés során, ha kovalens kötéseket létesít. Az elektronegativitása 2,5, ami egy közepes érték, a legtöbb atommal képes kovalens kötést létrehozni. A szénvegyületek molekulái általában diszkrét molekulák, nem ionosak és kevéssé polárisak. A szénatomnak, lévén a második periódusban foglal helyet, nincsenek alacsony energiaszintű d-pályái, ennek következtében nem csak a kötésben résztvevő elektronjainak száma, hanem a koordinációs száma is négy.
A szerves vegyületek jellegzetes fizikai tulajdonságokkal rendelkeznek:
-
a legtöbb szerves vegyület molekularácsban kristályosodik, a kicsi rácsenergia miatt az olvadáspontjuk általában alacsony (< 300 °C),
-
számos szerves vegyület illékony és bomlás nélkül desztillálható,
-
a szerves vegyületek zöme az elektromos áramot rosszul vezeti,
-
sok szerves vegyület jól oldódik különböző – szerves – oldószerekben.
Mielőtt a szénvegyületekben előforduló kötéseket tárgyalnánk, tekintsük át általánosan a kovalens kötést, amely leírására számos elmélet született. Ezek egyes részeit ma is alkalmazzuk.
A kovalens kötés egyszerű leírása
Lewis, Langmuir és Kossel elmélete szerint a kötést létrehozó atomok legkülső elektronhéjukon egy, vagy több elektronpárt egymással megosztanak, és ezzel kialakítják a nemesgázok elektronszerkezetét. Az első periódus esetében ez a héliumnak megfelelő, a második periódusban a neonnak megfelelő elektronszerkezetet. Mivel az utóbbi esetben ez 8 elektront jelent, innen származik az „oktett elmélet” elnevezés. A szerkezeti képletekben megrajzolt vegyértékvonalak egy-egy megosztott elektronpárt jelentenek.
A kötés létrejöhet a két összekapcsolódó atom egy-egy elektronjának megosztásával (kolligáció), vagy az elektronpár származhat csak az egyik atomtól (koordináció). Az elektronpár két atommag erőterében mozog, mindkettőhöz egyidejűleg tartozik, aminek következtében a két atommag távolsága kisebb, mint az atomok sugarainak összege, energiája pedig kisebb, mint a két diszkrét atomé.
Ez az elmélet ugyan könnyen alkalmazható, de nem vezethető le belőle pl. a molekulák valódi térszerkezete, nem ad választ a többszörös és delokalizált kötések kialakulására, paramágneses molekulák létezésére, stb.
A kovalens kötés leírása az atompályák segítségével:
A kovalens kötés korszerű leírásához szükség volt a kvantummechanika kifejlődésére, az atomok elektronhéjának felépülését kvantummechanikai alapokon tárgyaló megközelítésre. De Broglie anyaghullám elmélete 1924-ben az anyagi részecskék, köztük az elektron hullámtermészetét megalapozta, amelyekre alkalmazható a Schrödinger egyenlet. Ennek az egyenletnek a hidrogén esetében egzakt megoldása van, más esetekben közelítő megoldások vannak, az ún. az úgynevezett ψ = ψ (x, y, z) hullámfüggvények. A ψ amplitudó négyzete [ψ]2 arányos az elektron tartózkodási valószínűségével. Az azonos ψ értékű helyek egy burkolófelületeket adnak. Atompályának, vagy atomorbitálnak (AO), azt a burkolófelületet tekintjük, ahol az elektron 90%-os valószínűséggel megtalálható. Az atomokban minden elektron 4 kvantumszámmal jellemezhető: a fő-, mellék-, mágneses- és spinkvantumszámmal. Ismert, hogy a mellékkvantumszám szabja meg a pálya alakját. Az s-pályák gömbszimmetrikusak, a p-pályák súlyzó alakúak, a d-pályák alakja bonyolultabb.
|

|
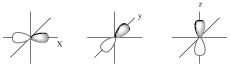
|
|
s-pálya |
p-pályák |
A vegyértékkötés elmélet (VB, valence bond)
Gyakorlatilag az elektronpár elmélet kvantummechanikai megközelítése. A molekula kialakulását, a kovalens kötés létrejöttét két atomorbitál átlapolásával írja le, ahol az azonos előjelű pályarészek lapolnak át.
A vegyértékkötés módszer szerint tehát kémiai kötés csak akkor jön létre két atom között, ha azok megfelelő atompályái képesek egymással átlapolni. A VB módszer lényege, hogy olyan hullámfüggvényekkel leírható elektronpárokat képez, amelyek megengedik, hogy a párt alkotó elektronok bármelyik atommag környezetében megtalálhatók legyenek. A kovalens kötés tehát úgy alakul ki a két atom között, hogy a két elektron nagy valószínűséggel tartózkodik a két atommag között, ami által egymáshoz köti azokat. A kötő állapotot leíró hullámfüggvény pedig azt mutatja, hogy két hullámfüggvény szuperpozíciója a megoldás. A kötő állapot hullámfüggvényének négyzete adja meg, hogy az elektron pár mekkora valószínűséggel található meg a tér egy pontján.
Az átfedő atompályák különbözőek lehetnek. Az átfedő pályáknak két típusa van: σ- és π-kötés. σ-kötés jön létre s-s, s-p és p-p pályák átlapolásakor akkor, ha a két megosztott elektron pályái fej-fej átfedésben vannak. Ezek a kötések a kötéstengelyre nézve forgásszimmetrikusak.
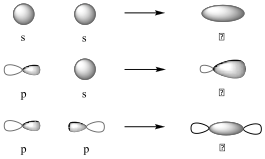
π-kötés keletkezik, ha két egymással párhuzamos pálya között van átfedés.
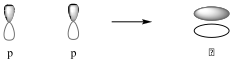
A σ-kötés körül szabad rotáció lehetséges, vagyis a kapcsolódó atomok elfordulhatnak, mivel az átlapolás mértéke ettől nem változik, míg a π-kötésnél a rotáció gátolt, az elfordulás az átlapolás csökkenésével, majd megszűnésével jár. Az elektronsűrűség a σ-kötésnél a kötéstengely körül a két mag között maximális, a π-kötésnél a kötés és a kötést létesítő p-atomorbitálok tengelyére merőleges sík alatt és felett maximális. A σ-kötés nehezen, a π-kötés könnyen polarizálható.
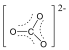
Vannak olyan ionok és molekulák, amelyeket ha a Lewis-képlettel írunk fel, több olyan szerkezet is lehetséges, ami csak elektronok helyzetében különbözik. Ilyen pl. az alábbi karbonátion is.
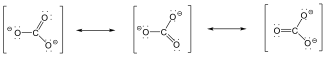
A karbonátionra három hipotetikus ún. mezomer határszerkezet írható fel, amelyek kombinációja írja le a valóságos szerkezetet. Az alapállapot, amelynek energiája kisebb, mint bármelyik szerkezeté, a szerkezetek kölcsönhatásaként adható meg, mely kölcsönhatást rezonanciának is nevezik.
A karbonátion esetében ez azt jelenti, hogy a három C-O kötés teljesen ekvivalens, a C-O kötéstávolság azonos.
A szerves kémiából választva példát, hasonlóak állapíthatók meg az acetát ionról.
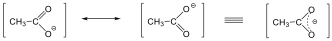
Általánosságban a mezomer határszerkezetekkel delokalizált π-rendszert írunk le. A mezomer határszerkezetek közé mindig a kétfejű nyilat használunk, nem összetévesztve az egyensúlyi folyamatok jelölésére használt kettős nyíllal. A kétfejű nyíl csak logikai szimbólum, nem azt jelenti, hogy a molekula elektroneloszlása e szélső állapotok között fluktuál.
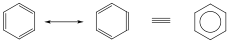
Az aromás vegyületek a gyűrűs delokalizált π-rendszert tartalmazó molekulák közé tartoznak, amelyekben a konjugált kettőskötésekből alakul ki a delokalizált π-rendszer. Legegyszerűbb képviselőjük a benzol.
Ha egy molekulába beépülő atom, vagy atomcsoport úgy kerül konjugációba az alapmolekulában lévő π-elektronrendszerével vagy p-pályával, hogy a teljes π-rendszer az új atom, vagy atomcsoport felé tolódik el, akkor ezt negatív mezomer effektust (-M) kifejtőnek tekintjük (pl. -NO2, -CN, -CHO, -COOH). Ennek ellentéte a pozitív mezomer effektus (+M), ilyenek pl. -F, -Cl, -Br, -I, -OH, -NH2.
A VB módszer és kiegészítései, pl. a rezonanciamodell, sok esetben lehetővé teszik a molekulák kötési energiájának kiszámítását, de az elektronpár elmélethez hasonlóan még több kérdésre nem ad választ.
A kovalens kötés leírása molekulapálya módszerrel (MO elmélete)
A MO módszer szerint a a molekulát felépítő atomok nem páronként lokalizált kötésekkel kapcsolódnak össze, hanem a többelektronos két- és többatomos molekula esetében - szigorúan véve - az összes elektron a molekulát alkotó atommagok erőterében mozog.
Elméleti megfontolások és tapasztalatok szerint azonban a molekulák szerkezetének leírásában néhány egyszerűsítés elvégezhető és a többatomos molekulák molekulaorbitáljai lényegében az érintett atomorbitálokból vezethetők le.
A molekulaorbitálok az alkotó atomorbitálok átlapolásával képződnek és betöltésük során az többelektronos atomok atomorbitáljainál megismert módon érvényesül a minimális energiára való törekvés és a Pauli-elv. A Pauli-elv érvényesülése azt jelenti, hogy az ellentétes spinű elektronok esetében energiaszegényebb, stabilabb állapot jöhet létre, mint azonos spinű elektronok esetében. Matematikailag ez az atompályák lineáris kombinációjával vezethető le.
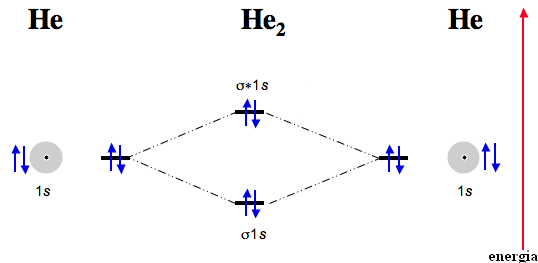
A H2 molekula esetében, a diszkrét H-atomok összekapcsolódásakor kialakuló kovalens kötésnél a H-atomok elektronjai egy közös un. kötő elektronpályára (σ) kerülnek, amelynek energiája kisebb, mint a diszkrét atomok AO energiája volt. Az atom orbitálnál nagyobb energiájú lazító (σ*) pályán nincs elektron.
A molekulák kémiai kötésének értelmezését megkönnyíti az AO-k és MO-k energiájánk, a rajtuk elhelyezjedő elektronok számának és spinjének ábrázolása, amint ez az alábbi ábrán a H2-molekula esetében látható:
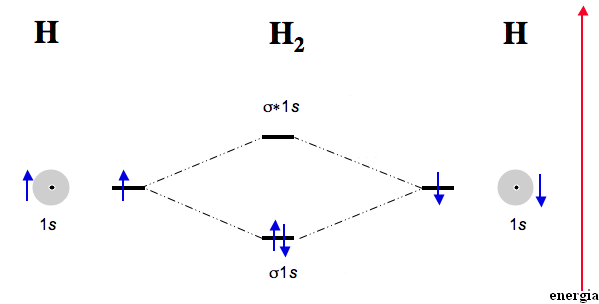
Általánosságban elmondható, hogy akkor alakul ki kötés, ha a kötő pályákon több elektron van, mint a lazító pályákon, ekkor jár a molekulakeletkezése energianyereséggel. Ezért nem létezik pl. a He2-molekula, mert esetében ugyanannyi elektron van a kötő pályán, mint a lazító pályán. A kötő és lazító pályákon lévő elektronpárok számának különbségét kötésrendnek nevezzük. A He2-molekula esetében a kötésrend 0, míg a H2-molekulánál 1.
A heterodinukleáris kovalens kötés kialakulásakor, pl. az alábbi esetben, amikor A és B atom létesít kovalens kötést, hasonlóan írható le a kötés kialakulása, annyi alapvető különbséggel, hogy A és B atom AO energiája nem azonos. A nagyobb rendszámú elemek atomorbitáljainak energiája kisebb, mint a kisebb rendszámú elemek azonos fő- és mellékkvantumszámú pályáinak energiája.
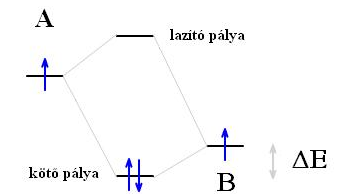
A képződő molekulapályák energiaszintje az atomorbitálok energiaszintjéhez képest szimmetrikusan helyezkedik el. A kötő pálya energiája pontosan annyival alacsonyabb érték a kisebb energiájú atomorbitálhoz képest, mint amennyivel a lazító pálya energiája magasabb a nagyobb energiájú atomorbitálnál.
Molekulaorbitálok nemcsak két s-, hanem két p- vagy egy-egy s- és p atomorbitál, stb. kombinációjával is létrejöhetnek. Kémiai kötés csakis azonos szimmetriatípusú AO-k kölcsönhatása révén létesülhet. Ha az s- és p-AO kombinációjával alakul ki kémiai kötés, ez csak akkor lehetséges, ha a px orientációjú atomorbitálról van szó. Két p- atomorbitál kombinációja révén nem jöhet létre kémiai kötés, ha a két AO merőlegesen orientált.
Különbség mutatkozik a különböző szimmetriájú atomorbitálok között létrejövő kötések tulajdonságaiban: σ-kötésnek nevezzük amikor kettő s- atomorbitál, vagy s- és px pályák között jönnek létre kötések. A két, a kötéstengelyre merőleges orientált py- (vagy pz-AO) kombinációjával létejövő kötéseket π-kötéseknek, a kötésben részt vevő elektronokat π-elektronoknak nevezzük. A σ-kötések kötési energiája azonos magtávolság esetén nagyobb a π-kötések kötési energiájánál.
A σ- és π-kötések esetében az eltérő szimmetria- és kötési energiaviszonyokon kívül további lényeges különbség van a rotáció szabadsága, valamint a bennük részt vevő elektronok polarizálhatósága tekintetében.
A szénatomnak alapállapotban az elektronszerkezete: 1s22s22px2py
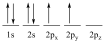
Ez alapján két kötőelektronnal rendelkezne. A tapasztalat szerint a legtöbb szerves vegyületben a szénatom 4 elektronjának felhasználásával létesít kapcsolatot: kettő, három vagy négy szomszédos atommal, vagyis 4 vegyértékű.
Szénatom négyes koordinációs számmal: amennyiben a szénatomhoz kapcsolódó 4 atom azonos minőségű (mint pl. a metánban), a négy kötés teljesen egyenértékű, a 4 atom egy tetraéder csúcsain foglal helyet. Ebből következik, hogy a kapcsolatokat létesítő négy kötőelektronpár azonos energiaszintű molekula pályákon (molekulaorbitálokon, MO) tartózkodik. Hogyan lehetséges ez?
Első lépésben egy ún. gerjesztés (promóció) következik be, aminek következtében a 2s pályán lévő elektronok közül az egyik az üres, magasabb energia szintű 2py pályára kerül. A 4 párosítatlan spinű elektron ezután pályakeveredés (hibridizáció) útján négy azonos energiájú hibridpálya jön létre, melyekben csak a pályák orientációja különbözik.
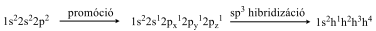
A létrejövő hibridpályák energiája sem sem az s-, sem a p-pályák energiájával nem azonos. A kialakuló hibridállapotot, mivel egy s- és 3 p-elektron részvételével jött létre, sp3 hibridállapotnak nevezzük. Ebben a 4 elektron azonos spinű, a hibridpályák térbelileg egymással 109,5°-os szöget zárnak be, tetraéderes elrendezésűek. Vegyületekben a négy hibridpálya a vele kötést kialakító atomok megfelelő atomorbitáljainak lineáris kombinációjával lokalizált, hengerszimmetrikus σ–kötést hoznak létre. A szénatom az ilyen vegyületekben sp3-hibridállapotban van. A C-C egyszeres kötés hossza: 154 pm
Szénatom hármas koordinációs számmal: azt az esetet, amikor a promócióval kialakult rendszerben a 2s-pályán elhelyezkedő elektron és 2 db p-pályán lévő elektron alakít ki hibridpályákat sp2 hibridizációnak nevezzük.
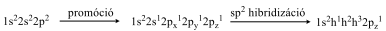
Ekkor a szénatom csak három további atomhoz fog kapcsolódni. A szénatom negyedik kötőelektronja a σ-kötések síkjára merőlegesen orientált p atomi pályán helyezkedik el és valamelyik szomszédos atom hasonló atomi pályáján lévő szabad kötő elektronnal létesít kötést. Így a két atom között még egy további kötés jön létre, tehát kettőskötés alakul ki. A kettőskötésben egy σ–kötés és egy π–kötés lesz. Az ilyen vegyületekben a kötésben lévő atomok egy síkban, egymáshoz képest 120°-os szöget bezárva helyezkednek el. Összességében a szénatomnak három szomszédos atomja lesz, melyekkel három σ-kötést és egy π–kötést alakít ki: az ilyen szénatomot nevezzük sp2 hibridállapotúnak. Az ilyen molekulákban a kötések nem egyenértékűek. Példák: etilén, H2CO. A C=C kettőskötés kötéshossza: 134 pm.
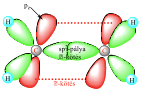
Az etilén kötései
Egy szerves molekula nem csak egy, hanem több C=C kettőskötést is tartalmazhat. Ha ezeket kettőskötéseket kettő vagy több egyszeres kötés választja el, vagyis izolált kettős kötésekről van szó, akkor a kettős kötések alig gyakorolnak egymásra hatást. Ha azonban a két kettőskötés között, legalábbis formailag egy darab egyes kötés található (konjugált rendszerek), akkor az ilyen vegyületeknél, a felváltva elhelyezkedő egyes és kettős kötések következtében, eltérő kötéshosszakat, új kémiai tulajdonságokat tapasztalunk. Pl. az 1,3 –butadiénben nem a C-C és C=C kötésekre jellemző 154 pm, illetve 134 pm értéket mérhetjük.
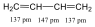
Tehát, konjugált rendszerekben az olefinkötések π-elektronrendszere befolyásolja a molekula elektroneloszlását. Ennek az az oka, hogy a sp2-hibridizált szénatomok közötti σ-kötések alkotta síkra merőlegesen helyezkednek el a pz-pályák, és a közbülső szénatomok pz-pályái érintkeznek a szomszédos szénatomokéval. Így, a formailag π-kötéssel kapcsolódó szénatomok között is kialakul az átfedésből σ-kötésnek tulajdonítható töltéssűrűség. A konjugáció következtében nem két-két atom közötti bicentrikus molekulapályák lesznek az ilyen molekulákban, hanem a konjugációban résztvevő atomok atompályáiból kialakuló multicentrikus pályák.
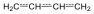
A konjugációban résztvevő elektronok alkotják a π-elektronhéjat. Ezen a héjon lévő elektronpárok nem lokalizálódnak atompárokhoz, hanem kettőnél több atommaghoz is tartoznak. A delokalizációval nem lesz ugyan egyenletes elektroneloszlás az egész molekulában, de a rendszer energiatartalmának csökkentésével (delokalizációs energia) növeli a termodinamikai stabilitását. A konjugációban nem csak szén, haem más atomok, pl. oxigén is részt vehet. Ilyen heterokonjugált rendszer pl. az akrolein.
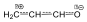
Szénatom kettes koordinációs számmal: ebben az esetben a promóciót követően egy, a 2s-pályán elhelyezkedő elektron és 1 db p-pályán lévő elektron alakít ki hibridpályákat, amit sp hibridizációnak nevezünk.
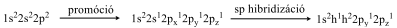
A szénatom így két σ-kötést tud létesíteni. Ez a két σ-kötés egy egyenes mentén helyezkedik el. A szénatom két fennmaradt kötőelektronja két, egymásra merőleges p atomorbitálon helyezkedik el és egyik, vagy mindkét szomszédos atom megfelelően orientált párosítatlan elektronjaival egy hármas, vagy két kettős kötést alakít ki.
Az acetilén kötései
Klasszikus példák: acetilén, HCN, RCN. A C≡C hármaskötés kötéshossza: 129 pm. A hármas kötésben lévő szénatomok egymáshoz közelebb helyezkednek el és erősebben is kötődnek, mint az egyszeres, vagy kétszeres C=C kötésben lévő szénatomok.
Az előzőekben részletezett hibridizációk jellemzőt az alábbi táblázatban foglaljuk össze:
Példák különböző hibridállapotú szénatomokra:
A benzol szerkezete
A benzol összegképlete: C6H6, szerkezete sokáig vitatéma volt. Ellentmondás volt ugyanis, hogy a kis C:H arány ellenére nem mutat telítetlen vegyületekre jellemző reakciókat, addíció nehezen játszódik le rajta és ellenáll az oxidációs reakcióknak is.
Kekule 1865-ben azt a mai is ismert szerkezeti formát, ahol a hattagú gyűrűben egyes és kettős kötések felváltva követik egymást.
A kvantummechanika, a kémiai kötés modern elméletének kidolgozásával a benzol pontos szerkezetének és reakciókészségének értelmezése is lehetővé vált. Műszeres mérésekkel megállapították, hogy a benzol molekulájában a hat szénatom és a hat hidrogénatom egy közös síkban foglal helyet. A szénatomok egy szabályos hatszöget alkotnak, minden C-C kötésnek azonos a hossza (139 pm), ami a Kekule-féle képlet szerint nem lehetséges.
A hat egyenértékű szénatom mindegyike rendelkezik egy, a gyűrű síkjára merőlegesen orientált p-orbitállal (pz), amelyeken 1-1 elektron tartózkodik. Ezek a p-orbitálok egymással átfedésben vannak, így a 6 db π-elektronnak a teljes gyűrűre kiterjedő delokalizációjára van lehetősége. A síkban elhelyezkedő szénvázat tehát mindkét oldalán egyenletes eloszlású π-elektronfelhő borítja. Ez a körkörös delokalizált -rendszer különleges stabilitást és kémiai tulajdonságokat biztosít a molekulának.

Az ilyen rendszereket aromásoknak nevezzük. Az ide tartozó vegyületek egy része a benzolból levezethető egy gyűrűt tartalmazó származék, de sok többgyűrűs, policiklusos vegyületet is ismerünk.
Hückel alkotta meg azokat a szabályokat, amelyek alapján eldönthető, hogy egy vegyület, függetlenül a vázát felépítő szénatokok számától, aromás-e. Ezek szerint a gyűrűnek síknak kell lennie, a gyűrű atomjai folytonos konjugációban vannak és a π–elektronok száma megfelel a 4n+2 értéknek (2,6,10,14…), ahol n=egész szám.
A szerves vegyületek csoportosítása, funkciós csoportok, nómenklatúra.
A szerves vegyületeket többféle szempont szerint csoportosíthatjuk. Az egyik ilyen szempont lehet a vegyületek összetétele: a csak szenet és hidrogént tartalmazó vegyületeket szénhidrogéneknek nevezzük. Az összes többi szerves vegyület ún. hetero atomot tartalmaz. Csoportosíthatunk a kötések jellege szerint is: a telített vegyületekben a szénatomok között csak egyszeres kötések vannak, míg a telítetlenekben a szénatomok között kettős és hármas kötések találhatók. A következő fejezetben a régebbi, klasszikusnak nevezhető váz szerinti rendszerezést, majd az újabb, funkciós csoport szerinti csoportosítást tekintjük át.
A szerves vegyületek elnevezése alapvetően kétféle úton történik. Használjuk a triviális és a szisztematikus elnevezéseket.
Triviális nevek: a vegyület valamilyen tulajdonságára, eredetére, stb. utal, véletlenszerűen alakult ki, a szerkezetre, az összetételre nem ad felvilágosítást. Pl. az ismert metanol triviális neve faszesz, mert korábban a fa száraz lepárlásával nyerték.
Ezzel szemben a szisztematikus nevezéktan (nómenklatúra) az elnevezés és a szerkezet között egyértelmű kapcsolatot teremt. 1892-ben Genfben történt az első nemzetközi szintű szabályozás kidolgozása és elfogadása a szerves vegyületek elnevezésének egységesítésére, hogy egy vegyületnek egyetlen neve legyen. Ezeket a szabályokat később sokszor módosították. Ma ezzel a kérdéssel a IUPAC (International Union of Pure and Applied Chemistry), a nemzetközi kémiai szervezet foglalkozik, ennek az irányelveit kell használni. Az angol és magyar elnevezéseknél, írásmódnál azonban eltérések fordulhatnak elő, a magyar szaknyelvben a Magyar Kémiai Elnevezés és Helyesírás Szabályaiban foglaltakat kell betartani.
A triviális nevek mellett ún. szubsztitúciós és csoportfunkciós elnevezéseket használunk.
A csoportfunkciós elnevezés alkalmazásakor kiválasztjuk a funkciós csoportnevet és a hozzákapcsoljuk a csoportok neveit, nincsenek elő- és utótagok. Leginkább az egyszerű, aciklusos halogéntartalmú vegyületek, alkoholok, éterek oxovegyületek esetén alkalmazható.
A szubsztitúciós nevezéktan szerint az elnevezni kívánt vegyületet egy alapvegyület helyettesített származékának tekintjük. Az alapvegyületet szigorú szabályok szerint választjuk ki, figyelembe kell venni, hogy milyen a vegyület szerkezete, milyen láncokat, gyűrűket stb. tartalmaz. Szükség esetén a vegyületben lévő szénatomokat rögzített elvek szerint megszámozzuk, meghatározzuk, hogy az egyes funkciós csoportokat elő-, vagy utótagként illeszthetjük-e a névhez.
A közismert CH3CH2-OH példáján mutatjuk be a kétféle elnevezést. A szubsztitúciós nómenklatúra alapján az alapvegyület az etán, ehhez illesztjük az alkoholok esetén használatos –ol végződést, így a név etanol lesz. A csoportfunkciós elnevezés szerint az etánból származtatott csoport az etil-, ehhez toldjuk az alkohol-t, így etil-alkohol lesz a neve.
Az egyes vegyületcsoportok részletes bemutatásánál minden esetben ismertetjük az elnevezési szabályokat, de a funkciós csoportok tárgyalásánál a legfontosabb elnevezések már ebben a fejezetben szerepelnek.
A vegyületek csoportosítása a vázuk szerint
A szerves molekulák váza lehet
-
nyíltláncú (nem elágazó és elágazó láncú)
-
gyűrűs (ciklusos).
A gyűrűs vegyületeket tovább rendszerezhetjük:
-
a gyűrűk száma (monociklusos, biciklusos, és policiklusos) szerint,
-
több gyűrűs vegyületek esetében a gyűrűk kapcsolódása szerint
-
izolált (nyíltláncú rész választja el a gyűrűket egymástól),
-
kondenzált (két gyűrűnek egy közös oldala van),
-
spiro (két összekapcsolódó gyűrűnek egy közös atomja van),
-
áthidalt (két összekapcsolt gyűrűnek több közös atomja van),
-
gyűrűtársulás (egy kötésen keresztül kapcsolódnak egymáshoz).
-
Ha a gyűrűt nem csak szénatomok építik fel, hanem más atomokat is tartalmaz, akkor heterociklusokról beszélünk. A karbociklusos (vagy izociklusos) vegyületekben a gyűrű valamennyi tagja szénatom.
Minden váz lehet telített, illetve telítetlen (kettős illetve hármas kötés(eke)t tartalmazó).
A monociklusos vegyületek és a kondenzált gyűrűrendszerek között lehetnek aromás vegyületek, amelyek vázában delokalizált π-rendszer található.
Néhány példa a fentiekre:
nyíltláncú, nem elágazó vegyületek:
|
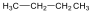
|
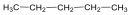
| |
|
n-bután |
n-pentán |
nyíltláncú, elágazó vegyületek:
|
|
| |
|
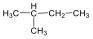
2-metil-bután |
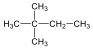
2,2-dimetil-bután |
gyűrűs vegyületek
|
|
|
|
|
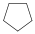
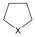
ciklopentán (telített) |
ciklohexén (telítetlen) |
benzol (aromás) |
A példákban szereplő valamennyi vegyület karbociklusos, mert a gyűrűk csak szénatomokból épülnek fel.
Monociklusos, biciklusos, és policiklusos gyűrűk:
|
|
|
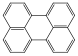
|
|
ciklopentán (monociklusos) |
naftalin (biciklusos, aromás) |
perilén (policiklusos) |
Példák izolált-, kondenzált gyűrűs és gyűrűtársulásos vegyületekre:
|
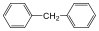
|
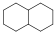
|
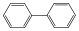
|
|
difenil-metán (izolált gyűrűs) |
biciklo[4.4.0]dekán (kondenzált gyűrűs) |
bifenil (gyűrűtársulás) |
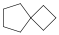
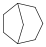
Példák spiro- és áthidalt gyűrűs vegyületekre:
|
|
|
|
spiro[3.4]oktán (spiro vegyület) |
biciklo[3.2.1]oktán (áthidalt gyűrűs) |
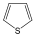
Példák heterociklusos vegyületekre:
|
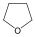
|
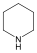
|
|
|
tetrahidrofurán (oxolán) |
piperidin |
tiofén |
Funkciós csoportok
Funkciós csoportnak a vázon helyet foglaló, legtöbbször heteroatomot/heteroatomokat tartalmazó csoportokat tekintjük, amelyek meghatározzák a vegyület kémiai tulajdonságait, a molekula reaktivitását. Ebben az értelemben a telítetlenségeket is a funkciós csoportok közé sorolhatjuk.
A funkciós csoportot tartalmazó vegyületeket a funkciós csoportot hordozó vázatom oxidációs állapota szerint célszerű csoportosítani. Azonos oxidációs állapotúnak tekintjük azokat a szénatomokat, amelyek azonos számú kötéssel kapcsolódnak szénnél elektronegatívabb atomhoz (pl. nitrogénhez, oxigénhez, kénhez, halogénekhez, stb.).
Legfontosabb vegyületcsaládok:
Alkánok (paraffinok): Telített szénhidrogének, nem tartalmaznak funkciós csoportot. Nevük végződése: „-án”.
Pl.: az első négy, a C1-C4 neve csak félig szisztematikus, a végződés szabályszerű: CH4, metán; H3C-CH3, etán; H3C-CH2-CH3, bután; H3C-CH2-CH2-CH3, majd a megfelelő görög számnév + –án utótag, pl. hexán. A belőlük formálisan egy hidrogén elvételével levezethető csoportok az alkilcsoportok, amelyek az –án helyett –il végződést kapnak, pl. CH4 → CH3- metil; H3C-CH3 → H3C-CH2- etil, stb..
Alkének: C=C kettős kötést tartalmazó szénhidrogének. Nevük végződése: „-én”.
Pl.: H2C=CH2, etén.
Alkinek: C≡C hármas kötést tartalmazó szénhidrogének. Nevük végződése: „-in”.
Pl.: HC≡CH etin, vagy a triviális név acetilén.
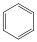
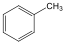
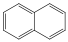
Aromás szénhidrogének: egy részük a benzolból levezethető származék, amely egy gyűrűt tartalmaz, de sok a bi-, tri… policiklusos vegyület is ismert.
|
|
|
|
|
benzol |
metil-benzol v. toluol |
naftalin |
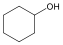
Alkoholok: hidroxilcsoportot tartalmazó vegyületek. Nevüket vagy a csoportfunkciós, vagy a szubsztitúciós nevezéktan szerint képezhetjük (pl. CH3OH, metil-alkohol, illetve metanol).
|
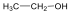
|
|
|
etanol v. etil-alkohol |
ciklohexanol v. ciklohexil-alkohol |
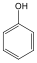
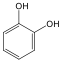
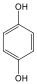
Fenolok: Az aromás gyűrűhöz kapcsolódó hidroxilcsoportokat tartalmazó vegyületeket fenoloknak nevezzük.
|
|
|
|
|
fenol |
1,2-dihidroxi-benzol pirokatechin |
1,4-dihidroxi-benzol, hidrokinon |
Éterek: Az éterekben az oxigénatomhoz két szénhidrogéncsoport kapcsolódik, tehát C-O-C kötést tartalmaz. Nevüket vagy a csoportfunkciós, vagy a szubsztitúciós nevezéktan szerint képezhetjük (pl. CH3-O-CH2CH3, etil-metil-éter, illetve metoxietán).
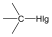
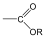
Halogenidek: A halogenidekben a szénvázhoz halogénatom kapcsolódik. Nevüket vagy a csoportfunkciós, vagy a szubsztitúciós nevezéktan szerint képezhetjük (pl. CH3Cl, metil-klorid, illetve klór-metán).
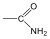
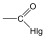
Aminok: Azokat a vegyületeket, amelyekben a szénvázhoz aminocsoportok kapcsolódnak, aminoknak nevezzük. Nevüket vagy a csoportfunkciós , vagy a szubsztitúciós nevezéktan szerint képezhetjük (pl. CH3NH2, metil-amin, illetve metán-amin).
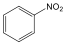
Nitrovegyületek: A nitrocsoportot (-NO2) tartalmazó vegyületek tartoznak ebbe a csoportba. Nevüket kizárólag a szubsztitúciós nómenklatúra szerint képezzük, a megfelelő szénhidrogén neve elé tett nitro előtaggal.
|
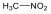
|
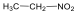
|
|
|
nitro-metán |
nitro-etán |
nitro-benzol |
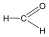
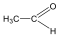
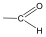
Aldehidek: Az oxovegyületek egyik csoportja, ahol a karbonilcsoporthoz (>C=O) egy hidrogénatom és egy alkilcsoport kapcsolódik. A legegyszerűbb aldehid a formaldehid, ami az alkilcsoport helyett is hidrogént tartalmaz. Az aldehideket elnevezhetjük aldehid végződéssel, a megfelelő karbonsav latin, vagy néhány esetben magyar nevéhez hozzátéve. illetve a szubsztitúciós nevezéktan szerint az „–al” végződés használatával; az általános név így az alkanal.
|
|
|
|
formaldehid v. metanal |
acetaldehid v. etanal |
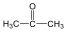
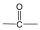
Ketonok: Az oxovegyületek másik nagy csoportja, ahol a karbonilcsoporthoz két alkilcsoport kapcsolódik. A legegyszerűbb keton az aceton. Nevüket vagy a csoportfunkciós elnevezési módszerrel keton utótaggal, vagy a szubsztitúciós nevezéktan szerint képezhetjük „-on” végződés használatával
|
|
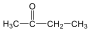
|
|
aceton v. propanon v.dimetil-keton |
etil-metil-keton v. bután-2-on |
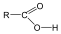
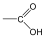
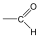
Karbonsavaknak nevezzük azokat a vegyületeket, amelyekben egy szénatomhoz kettős kötéssel egy oxigénatom és egyes kötéssel egy hidroxilcsoport kapcsolódik, a szénatom maradék egy vegyértékét pedig hidrogén, vagy alkilcsoport foglalja el.
|
R: |
|
|
| H metánsav(hangyasav) CH3 etánsav (ecetsav) |
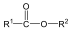
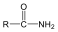
A karbonsavszármazékok közös vonása, hogy a karboxilcsoportban az oxigént és/vagy hidroxilcsoportot heteroatommal, csoport(ok)kal helyettesítjük.
|
|
|
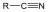
|
|
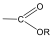
karbonsavészter |
karbonsavamid |
karbonsavnitril |
|
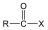 (X:halogén) |
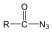
|
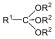
|
|
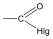
karbonsavhalogenid |
karbonsavazid |
ortoészter |
Az alábbi táblázatban megegyszer összefoglaljuk a legfontosabb funkciós csoportokat
|
alkán |
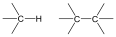
|
amin |
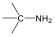
|
|
alkén |
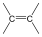
|
nitrovegyület |
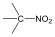
|
|
alkin |
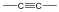
|
aldehid |
|
|
aromás |

|
keton |
|
|
halogénezett szh. |
|
karbonsav |
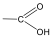
|
|
alkohol |
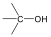
|
karbonsavanhidrid |
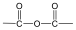
|
|
fenol |
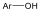
|
karbonsavészter |
|
|
éter |
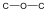
|
karbonsavhalogenid |
|
|
tiol |
|
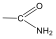
karbonsavamid |
|
|
tioéter |
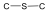
|
karbonsvanitril |
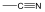
|
|
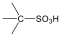
szulfonsav |
|
heterogyűrűs |
|
A szerves reakciók típusai, reakciómechanizmusok
A kémiai reakció során molekuláris szinten történik változás, kötések szakadnak fel és alakulnak ki újak elektronok átadásával, illetve felvételével, amit a reakciók mechanizmusa ír le. A reakciópartnerek egyike a reagens (reaktáns), a másik pedig a szubsztrát, amely valamilyen köztiterméken, esetleg köztitermékeken keresztül alakul át termékké, termékekké.
szubsztrát + reaktáns → [köztitermék(ek)] → termék(ek)
Megkülönböztetésük megállapodás szerint történik. Reagens az a komponens, amelynek a reakcióban részt vevő centruma heteroatom (nem szénatom), a szubsztrát pedig az, amelyiknek szénatomján a reakció végbe megy. Amennyiben mindkét reakciócentrum szénatom, akkor a reagens–szubsztrát fogalmak megválasztása önkényes, de általában a nukleofil partnert tekintjük reagensnek.
A szerves kémiai reakciókat több szempont alapján is szoktuk csoportosítani:
-
a kötések felszakadásának módja szerint;
-
egyetlen átmeneti állapoton keresztül, vagy köztitermékeken keresztül mennek-e végbe;
-
a kiindulási anyag és a termék szerkezetében lévő különbség szerint, vagyis milyen változással járt a reakció.
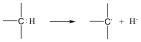
A szerves kémiai reakciók egy részében közvetlenül, egy lépésben alakulnak át az anyagok, de sokkal nagyobb számban ismerünk közbenső termékeken keresztül, több lépésben játszódó folyamatokat. Első lépésben a kovalens kötés felhasadása következik be, mely két úton lehetséges: a kötőelektronpár vagy szimmetrikusan, vagy aszimmetrikusan oszlik majd el a keletkező részecskék között. Azt az esetet, amikor az elektronok szimmetrikusan oszlanak el, homolitikus kötésfelhasadásnak, a másikat pedig heterolitikus kötésfelhasadásnak nevezzük.
Homolitikus kötésfelhasadás esetén a kötésben résztvevő kötőelektronpár egyenletesen oszlik meg a szétváló két részecske között és így semleges, párosítatlan elektronnal rendelkező gyökök keletkeznek. Ebben az esetben gyökös reakciókról beszélünk. Heterolitikus kötésfelhasadás esetén, attól függően, hogy a reagens milyen tulajdonságokkal rendelkezik és hogyan vesz részt a reakcióban két csoportot különböztetünk meg: nukleofil (magot kedvelő) és elektrofil (elektront kedvelő).
Egy C-H kötés többféleképpen is felhasadhat, a homolitikus út két gyök képződéséhez vezet.
A heterolitikus kötésfelhasadásnak további két változata lehet attól függően, hogy a kötőelektronpár melyik atomon marad. Egyik esetben a szénatomon marad mindkét elektron és karbanion, valamint hidrogénion képződik (szabad proton mindig valamilyen nukleofil ágenshez kötődve fordul elő, szabadon nem, pl. vizes oldatban hidroxónium-ion, H3O+ formájában), de az egyszerűség kedvéért a H+ jelölést alkalmazzuk):
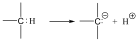
A másik esetben a hidrogénhez kerül az elektronpár és karbokation, valamint hidridion képződik. Karbéniumionnak azokat az ionokat nevezzük, amelyben a szénatom sp2 hibridállapotú.
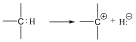
Homolízis nagyobb valószínűséggel következik be apoláris, vagy kevésbé poláris kovalens kötések esetében, míg a heterolízis az erősebben polározott kötésekre jellemző.
Homolízis, vagy heterolízis nem csak egyszeres, hanem többszörös kötéseken is bekövetkezhet.
A reakciókat, eredményük szerint négy fő típusba szokás sorolni: szubsztitúció, addíció, elimináció és izomerizáció (átrendeződés). Minden más reakció ennek a 4 típusnak valamilyen kombinációja, mint pl. a kondenzáció.
|

|
|
szubsztitúció |
|
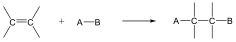
|
|
addíció |
|
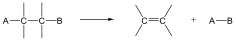
|
|
elimináció |
|

|
|
izomerizáció |
Szubsztitúciós reakciók:
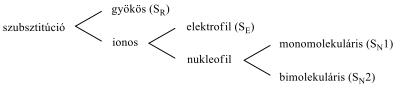
A szubsztitúciós reakció az egyszeres kötés felhasadásával és új egyszeres kötés kialakulásával játszódik le. A kilépő atom(csoport) helyére egy másik atom(csoport) lép be. Jele: S, a jobb alsó indexben pedig a mechanizmust tüntetjük fel, pl.: SR, a gyökös szubsztitúciót, SE, az elektrofil, SN a nukleofil szubsztitúciót jelöli.
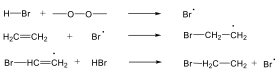
Sok szubsztitúciós reakció gyökös mechanizmus szerint játszódik le, amelyre példa szénhidrogének halogénezése. A metán klórozásakor a C-H kötés felhasításához 414 kJ/mól energia szükséges, míg a klór molekulában a kötési energia 243 kJ/mól, tehát a kisebb kötésenergiájú kötés fog első lépésben felhasadni.
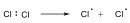
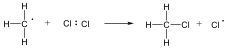
Következő lépésben a klór gyök leszakít egy hidrogént a metán molekuláról és sósav, illetve egy metilgyök képződik.

A metilgyök, ha klórmolekulával találkozik, metil-klorid képződése mellett egy klór gyök generálódik, ami az előzőek szerint tovább reagálhat.
Azokat az elemi lépéseket, amelyekben gyök képződik, fenntartják a láncreakciót, láncvivő lépésnek nevezzük.
Amikor két gyök találkozik, rekombináció történik, újabb gyök nem generálódik, a folyamat lánczáró lépés.
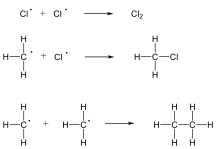
A gyökös mechanizmussal lejátszódó reakció legfőbb jellemzői: apoláris közegben, sokszor gázfázisban játszódik le; a reakció kezdőlépése egy homolitikus kötéshasadás, amit labilis paramágneses gyök vagy atom is kiválthat; a fény és oxigén befolyásolhatja a reakciót.
Az ionos mechanizmussal lejátszódó szubsztitúció lehet nukleofil és elektrofil, attól függően, hogy hogy a reagens elektronleadásra képes, "nukleofil" jellegű, vagyis a szubsztrátum elektronszegény (az atommagokhoz közeli) helyein támad (pl. a Lewis-bázisok), vagy elektronfelvételre hajlamos, "elektrofil" jellegű, tehát a szubsztrátum elektrondús helyein támad (pl. a Lewis-savak). Eszerint megkülönböztetünk nukleofil és elektrofil reakciókat.
A nukleofil reakciókat jobb alsó indexben N, az elektrofileket E indexekkel jelöljük, pl. a elektrofil szubsztitúció SE.
Az egymást követő (konszekutív) reakcióknál mindig a leglassabb, a reakció sebességét meghatározó elemi lépés jellegét vesszük figyelembe.
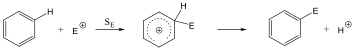
Az elektrofil szubsztitúciós reakcióra tipikus példa a benzol brómozása FeBr3 katalizátor jelenlétében:
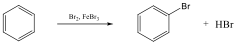
Első lépésben a katalizátor hatására kialakult az erősen elektrofil kation (E+), ami támad az aromás gyűrűn. Az előző példában így Br+ lesz az elektrofil ágens. Ha nem szimmetrikus az elektroneloszlás, akkor az elektronban gazdag helyen következik be a támadás.

Következő lépésben ez a kation addícionálódik az aromás gyűrűre, kialakul egy karbokation, ami aztán ledob egy protont és szubsztituált aromás vegyület alakul ki.
A nukleofil szubsztitúció egyik legjellegzetesebb példája lehet az alkil-halogenidek reakciója nukleofil ágensekkel.
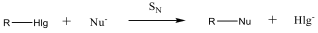
Nu-: F-. Cl-, Br-, I-, OH-, OR-, SH-, SR-, CN-, RCOO-, NH3
Mivel a szén-halogén kötés polarizált, az α–szénatom körüli elektronsűrűség kicsi, ez lesz a nukleofil ágens támadási pontja.
A nukleofil szubsztitúció molekularitása
Megkülönböztetjük a reakciókat aszerint, hogy a sebességmeghatározó lépésben hány molekula változtatja meg kötésállapotát. Monomolekuláris a reakció, ha a sebességmeghatározó lépésben csak a szubsztrátum vesz részt, bimolekuláris, ha a reagens is résztvesz. A kétféle reakcióútnak az alábbi legfontosabb jellemzői vannak:
Az SN1 reakció monomolekuláris, kétlépéses folyamat. Azért monomolekuláris, mert az első lépésben egy részecskén következik be kötésfelhasadás. Ennek során egy karbéniumion keletkezik, ami aztán reagálhat a nukleofil ágenssel.
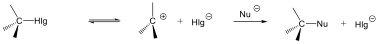
Az SN2 reakció ezzel szemben egylépéses, de ebben az egy lépésben mindkét reagáló eészecskén bekövetkezik kötésfelhasadás, ezért a reakció bimolekuláris.
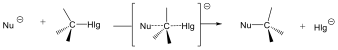
Összefoglalva a szubsztitúciós reakciókat, az alábbiak szerint csoportosíthatjuk:
Addíciós reakciók
Az addíciós reakció során egy többszörös kötéseket tartalmazó molekulába új csoportok épülnek be, miközben az eredeti π-kötés megszűnésével új σ-kötések alakulnak ki. Az addíciós reakciók jele: A, a jobb alsó indexben itt a mechanizmust tüntetjük fel, pl. AR, a gyökös addíciót, AN a nukleofil addíciót jelenti.
Gyökös addíció szerint játszódik le pl. olefinekre történő HBr addíció peroxid jelenlétében.
Elektrofil addíciós reakció az olefinekre történő halogénaddíció, amely peroxidmentes, poláris oldószerekben játszódik le.

Nukleofil addíciós reakció az oxovegyületek egyik legtipikusabb átalakulása, amelyben a nukleofil ágens a C=O polározott kettőskötésben elektronszegénnyé váló szénatomon hajt végre támadást és alakul ki addukt.
Az addíciós reakciók csoportosítása.
Eliminációs reakciók
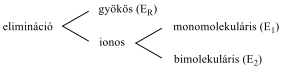
Az eliminációs (E) reakció lényegében az addíció megfordításaként értelmezhető: két π-kötés helyett egy σ-kötés alakul ki. Másképp megfogalmazva:
Az eliminációs reakciók jele: E, mellette a reakció molekularitását tüntetjük fel. A folyamat lehet mono- (E1) és bimolekuláris (E2)
Példa az eliminációs reakciókra az alkánok dehidrogénezése, a vicinális dihalogének dehalogénezése, vagy az alkoholok dehidratációja.
Telített szénhidrogénekből olefinek állíthatók elő gyökös eliminációs folyamatban:

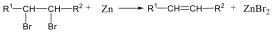
Olefinek előállíthatók vicinális dihalogén-származékból is cinkkel. A reakció is gyökös elimináció:
Alkoholokból vízelvonással is alkénekhez jutunk. A reakciót Lewis-savak (pl. AlCl3, ZnCl2) elősegítik:
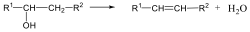
Az eliminációs reakciók csoportosítása:
Izomerizáció (átrendeződés) során a molekula összetétele nem változik, csak az atomok(csoportok) kapcsolódási sorrendje lesz más.
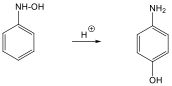
Példaként a N-fenil-hidroxil-amin Hofmann-Martius által elsőként leírt, tömény ásványi savak hatására bekövetkező átrendeződését mutatjuk be, amikor p-amino-fenol képződik.
Az egyes vegyületcsoportok tárgyalásakor még látunk példákat átrendeződésre, pl. a fenolok Fries-, a vicinális diolok pinakol-pinakolon átrendeződéseit.
A szerves vegyületek sav-bázis tulajdonságai
A reakciók értelmezéséhez nagyon fontos a szerves kémiában is a sav-bázis fogalmak ismerete és használata.
Az egyik legelső, jól használható sav-bázis elmélet Arrhenius nevéhez fűződik, aki savaknak tekintette azokat az anyagokat, amelyik vizes oldatban növelik annak hidrogénion koncentrációját, bázisoknak pedig azokat, amelyek a hidroxidionokét. Alapvető hiányossága ennek az elméletnek, hogy csak vizes oldatra definiált, más oldószerben, vagy gáz halmazállapotban nem használható.
A Brönsted-Lowry-féle értelmezés szerint savak azok az anyagok, amelyek protont képesek átadni egy másik anyagnak, míg a protonok felvételére képes anyagok a bázisok. Azok a folyamatok, amelyekben protonok átvitele történik a sav-bázis reakciók. Protont nem csak semleges molekula tud leadni, hanem anion, vagy kation is. A proton felvételére ugyancsak képes semleges molekula, anion, vagy akár kationok is.

Amikor a sav protont ad le, belőle a konjugált bázisa keletkezik, amely a protont újból felvéve visszaalakulhat savvá. Ilyen konjugált sav-bázis párok pl.: NH4+/NH3, CH3COOH/CH3COO-.
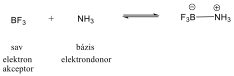
Ez az elmélet kibővítette ugyan a sav és bázis fogalmat, a sav-bázis reakciók tartományát, de csak protolitikus oldószerekben alkalmazható. Ezt a hiányosságot a Lewis-féle elmélet pótolta. A Lewis-féle felfogás szerint az elektronpárok befogadására képes ionokat és molekulákat savaknak, a elektronpár donorokat pedig bázisoknak kell tekinteni. Így tehát a savak koordinatívan telítetlen vegyületek, míg a bázisok nemkötő elekronpárokat tartalmaznak. Ezen besorolás szerint sav pl. az AlCl3, BF3, ZnCl2. Azok az anyagok, amelyek protonleadó képességük alapján savak, Lewis H-savaknak nevezte (pl. sósav). Az elmélettel jól értelmezhető sav-bazis rekcióként a BF3 és NH3 közötti reakció is.
Pearson a Lewis-féles savakat és bázisokat két-két csoportra bontotta, a savakon és bázisokon belül is megkülönböztette a soft (lágy) és hard (kemény) csoportokat. Az laza elektronhéjjal rendelkező, könnyen polarizálható és oxidálható, kis elektronegativitásúak kerültek a soft bázisok csoportjába, míg a nagyobb elektronegativitású, merev elektronhéjjal rendelkezők, nehezebben polarizálhatók és oxidálhatók az hard-ok közé. A soft savak akceptor atomjának relatíve nagy mérete és kis töltése van, laza az elektronhéja, amin könnyen gerjeszthető külső elektronok vannak (pl, ), míg az ezzel ellentétes tulajdonságúak a hard savak.
A sav-bázis fogalmakra a későbbiekben tárgyalandó vegyületcsoportok (pl. aminok, alkoholok, fenolok, stb.) aciditásának, bázicitásának értelemezésekor szükségünk lesz, ill. elméleti és gyakorlati szempontból is fontos a szerkezet és bázicitás/aciditás közötti összefüggés. Az aminok bázikus tulajdonsága ismert, ami a nitrogénatomjuk kötetlen elektronpárjának a következménye. Legpontosabban a szerkezet és bázicitás összefüggése gázfázisban tanulmányozható, mert ekkor nem kell pl. oldószerhatással számolni. Az említett aminok báziserősségében, viszonylagos bázicitásukban is jelentős különbség figyelhető meg pl. protikus és aprotikus oldószerekben.
A szerves vegyületek aciditásának-bázicitásának értelmezésekor több tényezőt is figyelembe kell venni. Ezek közül nagyon fontosan azok, amelyek a proton donor, vagy akceptor atom körüli elektroneloszlást befolyásolják, mert ez közvetlenül hat a donor-akceptor képességére.
Egyik ilyen effektus az ún. induktív effektus.
Az induktív effektus
A heteronukleáris kötéseknél, amikor a kötésben résztvevő atomok elektronegativitása eltérő, a kötések polarizáltsága figyelhető meg. A legfontosabbnak tekintett szén-heteroatom kötés polarizáltságának irányát és mértékét a két atom viszonylagos elektronegativitása határozza meg. Így a szén és halogénatomok közötti kötésekben az elektronfelhő a halogén atomok irányába tolódik el, az eltolódás mértéke pedig a C-F, -Cl, -Br, -I sorban csökken. A hatás nem korlátozódik csak egy kötésre, hanem csökkenő mértékkel, de hatással van a távolabb eső kötésekre is. Ha a C-H kötést vizsgáljuk, ezek is polarizáltak. Mivel itt a szénatomnak nagyobb az elektronegativitása, a kötésben résztvevő elektronok a szénatom irányába tolódnak el. Ezeket a hatásokat, amelyek a molekula alapállapotában elektroneltolódást okoznak, sztatikus induktív effektusnak (IS) nevezzük. A külső hatásra kialakuló eltolódásokat dinamikus effektusnak tekintjük. Sztatikus induktív effektust nem csak atomok fejthetnek ki, hanem atomcsoportok is. Az elektronfelhő eltolódási irányától függően különböztetünk meg negatív és pozitív induktív effektust. Az előző példában szereplő szén-halogén kötés esetén –IS, míg szén-fém kötésnél +IS effektus következik be. Az alkilcsoportok is +IS effektust fejtenek ki, ha elektronvonzó centrumhoz kapcsolódnak. Ellentétes a hatás, ha elektrontaszító centrumhoz kötődnek. A legtöbb atom, vagy atomcsoport –IS hatású, pozitív induktív effektust csak egyes negatív töltésű ionok (pl. COO-) és a kis elektronegativitású elemek (pl. P, Si, Sn, fémek) fejtenek ki.
Az induktív effektus mellett a mezomer elektroneltolódások is megfigyelhetők. A mezoméria fogalmát már bevezettük, amikor a mezomer határszerkezetekkel foglalkoztunk. Hangsúlyoztuk, hogy a felírható határszerkezetek formálisan elektronpárok elmozdulásával jellemezhetők, de semmiképpen sem reális elektroneltolódást jelentenek. Bizonyos esetekben azonban reális elektroneltolódással is kell számolnunk.
A mezomer elektroneltolódás
Az M-effektus akkor alakul ki, ha a hatást kifejtő atom vagy csoport közvetlenül egy telítetlen rendszerhez kapcsolódik. Ha a telítetlen rendszerhez kapcsolódó atom magános elektronpárral rendelkezik (pl. halogének), a hatást kifejtő csoport elektronsűrűsége csökken, a telítetlen rendszeré pedig nő: Ebben az esetben tekintjük pozitív előjelűnek a hatást (+M), mint a példaként szereplő vinil-klorid esetében.
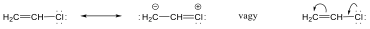
Az elektroneltolódás irányát görbe nyilakkal is szokták jelölni, előjelét ugyanúgy állapítjuk meg, mint az induktív effektusnál.
Amikor a telítetlen rendszerhez kapcsolódó atom(csoport) nem rendelkezik magános elektronpárral, ugyanakkor nagy elektronegativitású atom(ok)hoz kapcsolódik többszörös kötéssel (pl. formilcsoport), a heteronukleáris kötésen is bekövetkező elektroneltolódás miatt a telítetlen rendszer elektronsűrűsége csökken (-M).
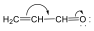
+M hatással rendelkező csoportok: -O-, halogének, -NR2, NH2, OH, OR, -SH, SR
-M hatással rendelkező csoportok: -NO2, COOH, COOR, -CONH2, -CN, -CHO, SO3H
A mezomer és induktív effektusok előjele lehet azonos vagy ellenkező. A hatások kombináltan jelentkeznek például az aromás SE reakcióra vonatkozó irányítási szabályoknál, amit az aromás vegyületek kémiai tulajdonságai között részletesen tárgyalunk.
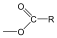
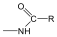
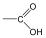
A legfontosabb funkciós csoportok induktív és mezomer effektusuk szerint csoportosítva:
|
+I, +M |
-I, +M |
-I, -M |
|
-CH3 -CH2R -CHR2 -CR3 -O- -S- |
halogének -OH, -OR -NH2, -NHR, NR2
|
-CF3 -NO2 -CN
|
Feladatok:
-
Jellemezze a kötő- és lazító elektronpárokat (energiaszint és tartózkodási valószínűség alapján)!
-
Mit értünk atom- és molekulaorbitál alatt?
-
Mi a molekulapálya-módszer alapelve? Mire alkalmazható?
-
A cellás ábrázolással adja meg a hidrogénmolekula elektronszerkezetét!
-
Mit jelent a szénatom rendűsége?
-
Mit jelent az sp3, sp2 hibridizáció?
-
Mi jellemzi a σ– és π–kötéseket?
-
Mit jelentenek a következő fogalmak?
a.) kötéshossz
b.) mezomer effektus
c.) homolitikus kötésfelhasadás
d.) gyök
-
Milyen reakciótípusokat ismer?
-
Jellemezze az alábbi reakciótípusokat és írjon rájuk példákat is 1-1 egyenlettel
a.) szubsztitúció
b.) addíció
c.) elimináció
-
Milyen funkciós csoportokat tartalmaznak az alábbi vegyületek?
Tartalom
A királis molekuláknak nincs belső szimmetriasíkjuk, ezért a molekula és tükörképi párja két eltérő térszerkezetnek felel meg. A tükörképi párokat más néven enantiomereknek nevezzük. A molekula kiralitáscentruma a tetraéderes hibridizációjú szénatom, amelyhez négy különböző szubsztituens kapcsolódik.
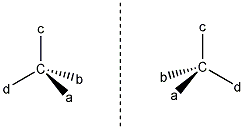
A térszerkezetek síkban való ábrázolásához legelterjedtebben projektív képleteket használnak. Ehhez E. Fischer javaslatára egységes vetítési szabályokat dolgoztak ki, amelynek a lényege a következő:
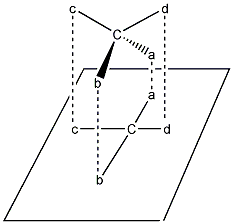
A királis szénatom köré elképzelhető tetraédert úgy orientáljuk, hogy a vetítési síkhoz közelebbi éle a síkkal párhuzamosan felülről lefelé haladó irányban helyezkedjen el, úgy, hogy a magasabb oxidációs fokú csoport legyen felül. Ezután a kapcsolódó csoportokat a síkra merőleges sugarakkal a síkra vetítjük. A projektív képleteken végzett páratlan számú szubsztituenscsere az ellenkező enantiomerhez vezet, a konfiguráció megváltozásával jár. Páros számú szubsztituenscsere nem változtatja meg a térszerkezetet.
Az enantiomerekben az atomcsoportok egymástól való távolságai egyformák, ezért a molekulán belüli és a molekulák közötti kölcsönhatások is azonosak, a két enantiomer energiatartalma megegyezik. Ez azonos kémiai és fizikai tulajdonságokat kölcsönöz a tükörképi pároknak, ezért fizikai, kémiai tulajdonságaik megegyeznek.
A két enantiomer csak valamilyen királis hatással szemben viselkedik különbözőképpen. Ilyen lehet pl. egy másik királis molekulával végbemenő reakció. A leggyakrabban alkalmazott királis hatás, amellyel a tükörképi párok között különbséget lehet tenni, a síkban poláros fény. A síkban poláros fény eltérő kölcsönhatásba lép a két enantiomerrel, és ez a rezgési sík elfordulását erdményezi. A jelenséget optikai aktivitásnak, az olyan vegyületeket pedig, amelyek elforgatják a síkban polarizált fény síkját, optikailag aktív vegyületeknek nevezzük.
Az optikailag aktív enantiomer vegyületpár egyik tagja ugyanolyan mértékben forgat jobbra, mint a másik balra. Az optikai aktivitást polariméterrel határozzák meg.
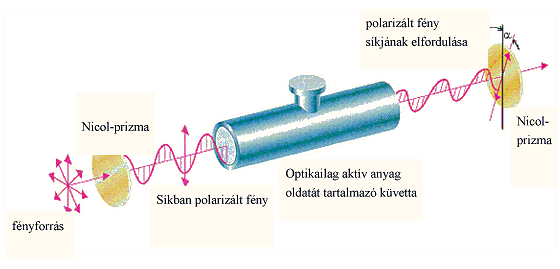
Azokat a molekulákat, amelyek a poláros fény síkját az óramutató járásával megegyező irányban forgatják el, jobbra forgatóknak (+), amelyek az óramutató járásával ellenkező irányban forgatnak, balra forgatónak (-) nevezik.
Ha az oldatban a két enantiomer azonos mennyiségben van jelen, oldatukat racém elegynek nevezzük, amely optikailag inaktív.
Az optikai forgatóképességet, a forgatás szögét -val jelöljük. A fajlagos forgatóképesség jelölése [], annak a fokokban mért szögnek a százszorosa, amellyel a poláros fény síkja elfordul, ha a vizsgált anyag 1 vegyes %-os oldatának 1 dm-es rétegén halad át.
A fajlagos forgatóképesség értéke függ az oldószertől, a hullámhossztól és a hőmérséklettől is. A hőmérsékletet (°C) felső indexként, a fény hullámhosszát alsó indexként adják meg (D: Na D-vonala: 589 nm).
![]()
: a poláros fény síkjának fokokban mért elfordulása
c: az oldat koncentrációja g/100 cm3 oldatban
l: az optikai úthossz dm-ben
A királis szénatom négy különböző szubsztituense kétféle térbeli elrendeződésben, két konfigurációban helyezkedhet el. A XIX. század végétől felmerült az igény, hogy az enantiomereket megkülönböztessék egymástól. Mivel akkor ehhez tudományosan megalapozott módszer nem állt rendelkezésre, E. Fischer önkényesen a glicerinaldehidben lévő királis szénatom konfigurációját javasolta viszonyítási alapnak.
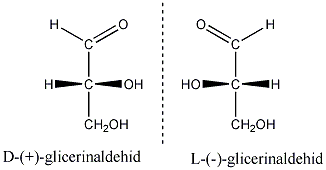
A D-glicerinaldehiddel szerkezeti rokonságban lévő vegyületek azok, amelyek konfigurációját olyan kémiai átalakításokkal vezetik vissza a D-glicerinaldehidre, amelyek a kiralitáscentrum konfigurációját nem módosítják. Ezt relatív konfigurációnak nevezik, és a D/L jelölés nincs összefüggésben a forgatásuk irányával.
Az 1950-es évek elején röntgendiffrakciós módszerrel igazolták, hogy a (+)-glicerinaldehid valós térszerkezete megegyezik a D-glicerinaldehidre önkényesen felrajzolt szerkezettel.
1956-ban Cahn, Ingold és Prelog olyan rendszert dolgoztak ki, amely alkalmas arra, hogy a molekula tényleges térszerkezetét vetített képlete alapján kiralitáscentrumának abszolút konfigurációját megadhassuk. Eljárásuk lényege, hogy a királis szénatomhoz kapcsolódó szubsztituenseknek adott szabályok szerint megállapítják a sorrendjét. Ennek bemutatásához a D-glicerinaldehid szerkezeti képletét használjuk fel.
1., A királis szénatom szubsztituenseit sorrendbe állítják. A legnagyobb rendszámú kapcsolódó atom kapja a legkisebb sorszámot, majd úgy haladnak a legkisebb rendszámú felé. Ha ezek között azonosakat találunk, pl. a –CHO és a –CH2OH csoportban is szénatom kapcsolódik a királis atomhoz, akkor az ezekhez kapcsolódó további atomokat vizsgáljuk. Mivel itt is oxigénatomok kapcsolódnak, ezért a ligandumok koordinációs számát négyre egészítjük ki és a többszörös kötésekben lévő atomokat megtöbbszörözzük úgy, hogy egyszeres kötések alakuljanak ki. Ennek alapján jön létre az alábbi rangsor:
-OH (1) > -CHO (2) > -CH2OH (3) > H (4)
2., Síkra vetített képleteknél úgy járunk el, hogy a legnagyobb sorszámú csoport kerüljön alulra, majd a szubsztituensek csökkenő prioritás szerinti sorrendjét vizsgálva azt mondhatjuk a molekuláról, hogy ha sorrendjük szerint a körbejárás iránya megegyezik az óramutató járásával, akkor R-, ha azzal ellentétes, akkor S-konfigurációjú.
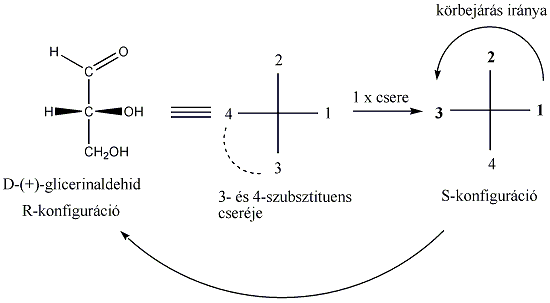
A fenti példán a körbejárás iránya az óramutató járásával ellentétes, de mivel a csoportok rendezésekor páratlan számú cserét hajtottunk végre, az az eredeti konfiguráció megváltozását eredményezte, így a D-glicerinaldehid R-konfigurációjú.
Számos olyan molekula található a természetben, amelyek több kiralitáscentrumot is tartalmaznak. Az sztereoizomerek száma a királis atomok számától függ: 2n, ahol n a királis atomok száma.
Így pl. a négy szénatomos, aldehidcsoportot tartalmazó szénhidrátokban két királis szénatom van, a lehetséges sztereoizomerjeik száma 22=4.
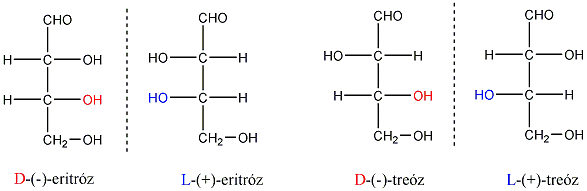
Látható, hogy a D- és L-eritróz, valamint a D- és L-treóz egymás tükörképei. Ugyanakkor az eritróz és a treóz egyik királis centrumának konfigurációja azonos, a másik különböző. Az olyan sztereoizomereket, amelyekben legalább egy királis centrum konfigurációja megegyező, ugyanakkor legalább egy királis centrum konfigurációja különböző, diasztereomereknek nevezzük. A diasztereomerek energiatartalma nem egyenlő, ezért fizikai és kémiai tulajdonságaik is különböznek egymástól.
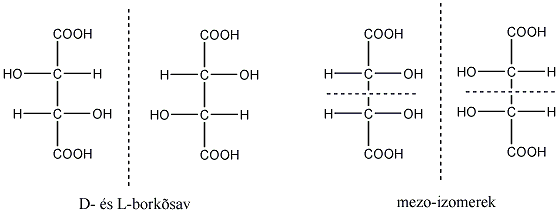
Előfordulnak olyan molekulák is, amelyekben a királis szénatomokhoz azonos csoportok kapcsolódnak, ilyen pl. a borkősav. A D- és L-borkősav két aszimmetriacentrumot tartalmaz, egymásnak tükörképi párjai. A mezo-izomerekben azonban van egy belső tükörsík, amire nézve a molekula két fele tükörképi párt alkot, ennek következtében a királis szénatomok azonos nagyságú, de ellentétes irányú forgatásai kiegyenlítik egymást. Ezért ezek a molekulák optikailag inaktívak, a poláros fény síkját nem forgatják el.
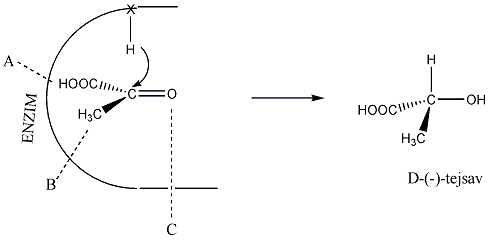
A természetes vegyületek között számos királis molekula található pl. a szteroidok, aminok, aminosavak, szénhidrátok, karbonsavak között. Számos biológiai reakcióban csak az egyik enantiomer keletkezik, vagy az egyik enantiomernek teljesen eltérő biológiai hatása van, mint a másiknak. A biológiai átalakulásokat koordináló enzimek válogatnak az enantiomerek között. Az enzim kiralitása az őt felépítő aminosavak kiralitásából fakad. Az alábbi ábra azt illusztrálja, hogy a piroszőlősavból egy enzimatikus redukció során csak az egyik enantiomer tejsav keletkezik, mert a piroszőlősav csak egyféleképpen tud az enzim aktív helyéhez kötődni:
1., Állapítsa meg, hogy a koleszterol molekula hány királis szénatomot tartalmaz. Mennyi a lehetséges sztereoizomerek száma?
2., Mi a D-tejsav abszolút konfigurációja?
3., Hány aszimmetriacentrum van az - és a -D-glükózban és ezek a molekulák milyen viszonyban vannak egymással?
Tartalom
- 7. Telített szénhidrogének
- 8. Telítetlen szénhidrogének: Alkének
- 9. Telítetlen szénhidrogének: Alkinok
- 10. Aromás szénhidrogének
Tartalom
A természetben nagy mennyiségben találhatók szénhidrogének a kőolajban és a földgázban. Kőolajfinomítókban az egyes komponenseket forráspont különbség alapján, frakcionált desztillációval szétválasztják.
Az alkánok elnevezésének tudományos rendszerét már 1892-ben elfogadták, kialakítva a szerves vegyületek elnevezésének alapelveit. A IUPAC által elfogadott rendszer alapelvei a következők:
-
A görög számnév tövéhez illesztett –án végződés adja meg a telített szénhidrogén nevét; kivételt képez a négy legkisebb szénatomszámú alkán neve, ezeknél meghagyták a régi eredetű triviális elnevezést.
-
Az alkán szerkezeti képletében megállapítjuk a leghosszabb nem elágazó szénláncot. Ez megadja a vegyület szisztematikus nevének alapját.
-
A leghosszabb folyamatos szénlánc szénatomjainak megszámozása olyan módon, hogy az elágazó szénatom a legkisebb számot kapja.
-
Az elágazó szénatomokon található szubsztituensek azonosítása. Az elnevezés alapjául szolgáló alkán neve elé illesztjük a megfelelő alkilcsoport nevét, számmal jelölve helyzetét. Több kapcsolódó szubsztituens esetén mindegyiknek megadjuk a nevét és helyzetét az elnevezés alapjául szolgáló normális alkánban.
-
Amikor két szubsztituens kapcsolódik ugyanazon szénatomhoz, akkor a szénatom számát többször megnevezzük.
-
Amikor a szubsztituens többször fordul elő a vegyületben, akkor a di-, tri-, tetra-, penta- stb. előtaggal jelöljük meg a mennyiségét, a számokat pedig vesszővel különítjük el egymástól.
A fenti legfontosabb alapelveket az alábbi ábra foglalja össze:
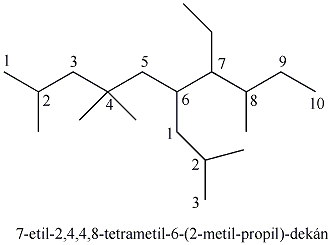
A molekulák egy tetszőleges részletét, az egymáshoz kapcsolódó atomok egy meghatározott együttesét csoportnak nevezzük. A csoport önmagában nem létezik, de alkalmazása hasznos a szerves vegyületek elnevezése, ill. a szerkezeti egységeinek elkülönítése szempontjából. Az alkánokból levezethető alkilcsoport az egyik láncvégi hidrogénatom elvételével jön létre. Nevét úgy lépezzük, hogy a megfelelő szénhidrogén nevének –án végződését –il végződésre változatjuk.
A telített szénhidrogénekben a tetraéderes térszerkezettel rendelkező sp3 hibridállapotú szénatomok kizárólag egyszeres kötésekkel kapcsolódnak egymáshoz.
A szénatomszám növekedésével a lehetséges izomerek száma rohamosan nő. A nem elágazó szénláncot normális szénláncnak nevezzük. A n-alkánok homológ sort alkotnak, melynek egymás utáni tagjai egy -CH2- (metilén) csoporttal különböznek egymástól.
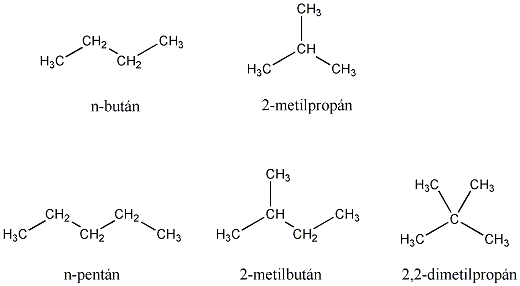
A négyszénatomos vagy annál hosszabb szénláncot tartalmazó alkánoknak többféle eltérő szerkezetű képviselője is lehet. Így pl. a C4H10 összegképletű alkánnak két képviselője lehet, a bután és a 2-metil-propán, ezek egymásnak szerkezeti izomerei. Ugyanígy a C5H12 összegképletű telített szénhidrogén lehetséges szerkezeti izomerei is felírhatók:
Az alábbi táblázat az első tíz normális szénláncú alkán nevét, szerkezeti képletét, a származtatott alkilcsoportot és a lehetséges szerkezeti izomerek számát mutatja be:
|
Név |
Összeg-képlet |
Normál szénláncú izomer szerkezeti képlete |
Alkilcsoport |
Szerkezeti izomerek száma |
|
metán |
CH4 |
CH4 |
CH3- metil |
1 |
|
etán |
C2H6 |
CH3-CH3 |
CH3-CH2- etil |
1 |
|
propán |
C3H8 |
CH3-CH2-CH3 |
CH3-CH2-CH2- propil |
1 |
|
bután |
C4H10 |
CH3-CH2-CH2-CH3 |
CH3-CH2-CH2-CH2- butil |
2 |
|
pentán |
C5H12 |
CH3-(CH2)3-CH3 |
CH3-(CH2)3-CH2- pentil |
3 |
|
hexán |
C6H14 |
CH3-(CH2)4-CH3 |
CH3-(CH2)4-CH2- hexil |
5 |
|
heptán |
C7H16 |
CH3-(CH2)5-CH3 |
CH3-(CH2)5-CH2- heptil |
9 |
|
oktán |
C8H18 |
CH3-(CH2)6-CH3 |
CH3-(CH2)6-CH2- oktil |
18 |
|
nonán |
C9H20 |
CH3-(CH2)7-CH3 |
CH3-(CH2)7-CH2- nonil |
35 |
|
dekán |
C10H22 |
CH3-(CH2)8-CH3 |
CH3-(CH2)8-CH2- decil |
75 |
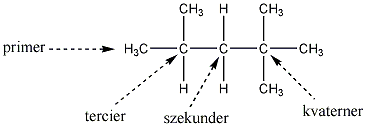
A több szénatomos vegyületek szénatomjai egymáshoz való kapcsolódásuk módja szerint négyféle strukturális helyzetben lehetnek. Primer, szekunder, tercier és kvaterner szénatomokat különböztetünk meg, attól függően, hogy hány másik szénatomhoz kapcsolódnak.
Az sp3 hibridizációjú szénatomok tetraéderes szerkezete a kapcsolódó atomok térbeli helyzetét alakítja ki. Ezért az alkánok pontos szerkezete csak térben adható meg, figyelembe véve a C-C egyes kötések körüli rotációt is. Az egymással közvetlenül nem kapcsolódó atomok térbeli viszonyát a konformáció írja le. Az etánmolekulának két szélsőséges konformációját lehet megkülönböztetni. A fedőállású szerkezetben a két metilcsoport hidrogénjei a lehető legközelebb vannak egymáshoz. Ha a modellt szemből nézzük, akkor a hidrogénatomok éppen fedik egymást. Amikor a hidrogénatomok között a távolság maximális, a konformert nyitott állásnak nevezik. A két szerkezet között a szénatomok közötti -kötés körüli szabad rotáció miatt végtelen sok más szerkezeti lehetőség is van. A nyitott állás szabadentalpia tartalma 11,7 kJ/mol értékkel alacsonyabb, mint a fedő állapotban lévő etánmolekuláé. Ez az energiakülönbség olyan csekély, hogy az atomok és atomcsoportok hőmozgásának kinetikai energiája ezt meghaladja, ezért a két konformer folyamatosan átalakul egymásba, egymástól nem különíthetők el. A konformereket fűrészbak diagrammal (ábra felső fele) vagy Newman projekcióval (ábra alsó része) ábrázolják:
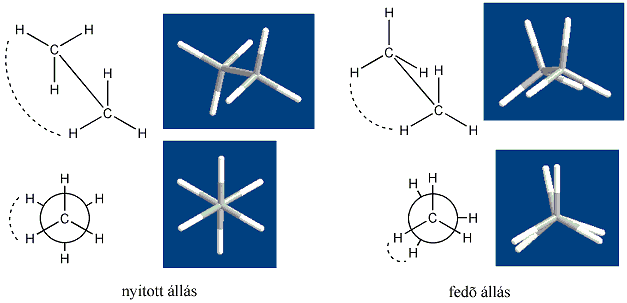
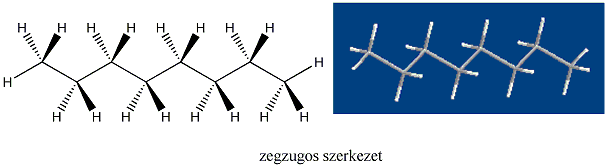
A hosszabb szénláncú paraffinok konformációja is hasonlóan értelmezhető, így nagyobb valószínűséggel a nyitott elhelyezkedésnek megfelelő zegzugos téralkat alakul ki.
A szénatomokból álló gyűrűt tartalmazó telített szénhidrogének a cikloalkánok. Általános képletük CnH2n. Nevük megegyezik az azonos szénatomszámú, normális láncú alkánokéval, csak a név elé a gyűrűre utaló ciklo szót tesszük.
A gyűrű szubsztituenseit a helyzetükre utaló számmal együtt a gyűrűs alapvegyület neve előtt adjuk meg.
A három- és négytagú gyűrűk esetén jelentős eltérés tapasztalható a tetraéderes szögtől (109,5°). A kialakuló szögfeszültség és az egymás mellett lévő -CH2- csoportok fedő állása miatt fellépő torziós feszültség a ciklopropánt és a ciklobutánt instabillá teszi.
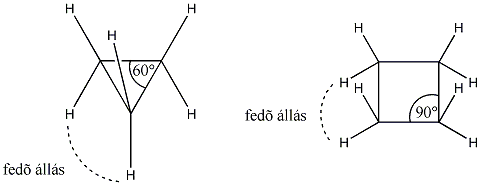
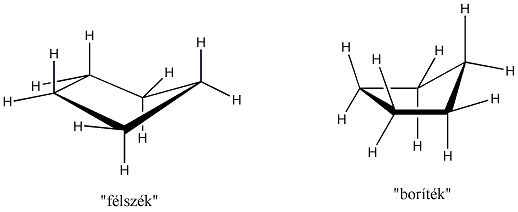
A ciklopentánban nincs szögfeszültség, de a C-H kötések fedő állása torziós feszültséggel jár. A gyűrű síkjának elcsavarodásával a torziós feszültség csökken, így „boríték” ill. „félszék” stabilis konformációk alakulnak ki.
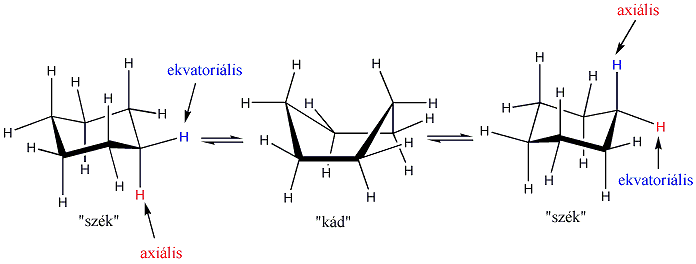
A ciklohexán gyűrűje teljesen feszültségmentes konformációt alakít ki azzal, hogy minden szénatom körül szabályos tetraéderes elrendeződés alakul ki és a szomszédos -CH2- csoportok is nyitott állásban vannak. Ez úgy valósulhat meg, hogy a gyűrű atomjai nem egy síkban helyezkednek el. Ez a „szék” konformer. A „kád” konformer kevésbé kedvező, mert a gyűrűt alkotó szénatomok közül négy esetében a hidrogénatomok fedő állásúak. A két konformer szobahőmérsékleten egymásba átalakul, egyik sem izolálható.
A ciklohexánban a szénatomokból kiinduló két-két C-H kötés közül az egyik a gyűrű síkjára merőlegesen (axiálisan) helyezkedik el, a másik iránya alig tér el a gyűrű síkjától (ekvatoriális). A ciklohexán konformerek gyors egymásba alakulása során az ekvatoriális kötésirány axiálisra módosul, és viszont.
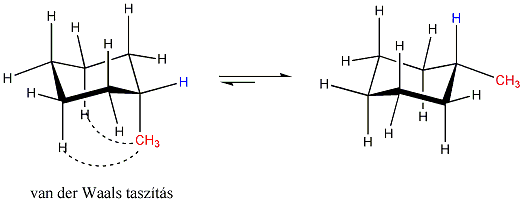
Amikor azonban a ciklohexángyűrűhöz valamilyen szubsztituens kapcsolódik, akkor a kétféle székkonformer nem azonos, a nagyobb tértöltésű szubsztituens lehetőleg ekvatoriális helyzetben található. Ennek az az oka, hogy az axiális helyzetű csoport közel kerül a C-3 és C-5 axiális hidrogénatomokhoz és van der Waals-taszítás lép fel:
Telítetlen vegyületek katalitikus hidrogénezése is alkánokat eredményez:
![]()
Zsírsavak nátrium sóját nátrium-hidroxid és kalcium-oxid jelenlétében hevítve alkánokhoz jutunk:
![]()
Ketonok cinamalgám jelenlétében végzett tömény sósavas redukciója alkánokat eredményez:
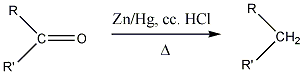
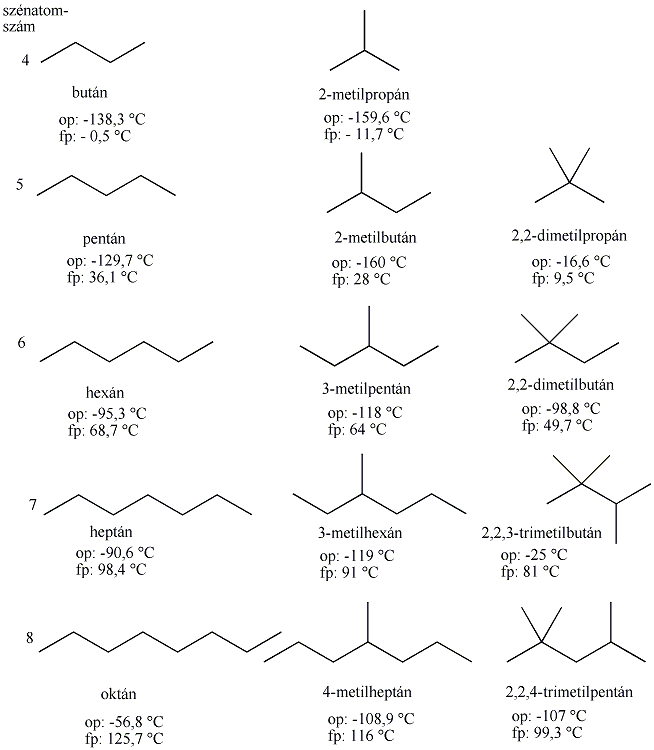
A normális szénláncú paraffinok homológ sorának első négy tagja szobahőmérsékleten gáz halmazállapotú, a C5-C16 alkánok folyékonyak, a többi pedig szilárd. Forráspontjuk a szénatomszám növekedésével nő. Az olvadáspont is emelkedik, de nem olyan szabályosan, mint a forráspont. Szabályosság akkor figyelhető meg, ha a páros és páratlan szénatomszámú alkánokat külön vizsgáljuk. Az eltérés abból adódik, hogy a páros és páratlan szénatomszámú alkánok eltérő kristályrendszerben kristályosodnak, az eltérő kristályalkatok rácsenergiája különböző, az olvadáspont pedig a rácsenergia függvénye.
Az elágazó láncú alkánok forráspontja kisebb, mint a megfelelő normális alkáné.
Az alkánok apoláros vegyületek, ezért vízben gyakorlatilag nem oldódnak.
Azonos szénatomszámú, normál és elágazó szénláncú alkánok olvadás és forráspontjának összehasonlítását a következő ábra szemlélteti:
Az alkánok és cikloalkánok stabilis vegyületek, erre utal a paraffin (parum affinis = csökkent reakciókészség) elnevezés is. Szobahőmérsékleten savakkal, lúgokkal nem reagálnak, oxidálószerekkel szemben is közömbösek.
Elterjedten használják őket energiahordozóként (tüzelőanyagként, motorhajtó anyagként), mert magasabb hőmérsékleten elégnek és a reakciót jelentős exoterm hőváltozás kíséri:
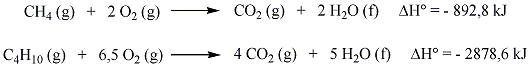
A tökéletes égés feltételei azonban ritkán teljesülnek ezekben a folyamatokban, az oxigén hiánya számos káros melléktermék – szén-monoxid (CO), korom (C), formaldehid (HCHO) és ecetsav (CH3COOH) – képződéséhez vezet.
Az alkánok hidrogénatomjai halogénekkel kicserélhetők. A halogénezés fény és/vagy hő hatására fluorral robbanásszerűen, klórral könnyen, brómmal lassabban játszódik le. Jóddal nem megy végbe a reakció.
A halogénezés gyökös mechanizmusú láncreakció, melyet a metán klórozása példáján szemléltetünk.
Megvilágítás hatására a klórmolekula homolitikusan bomlik, klórgyökök keletkeznek:
![]()
A reakcióképes klóratom a metánmolekulával ütközve abból egy hidrogénatomot hasít ki, így sósav és metilgyök keletkezik:
![]()
A metilgyök egy újabb klórmolekulával metil-kloridot és klórgyököt hoz létre, ezzel a reakció továbbhalad:
![]()
A lánczáró lépésekben nem keletkezik újabb gyök:
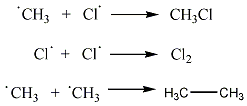
A metán klórozása di-, tri-, tetraklór származék képződéséhez is vezethet:
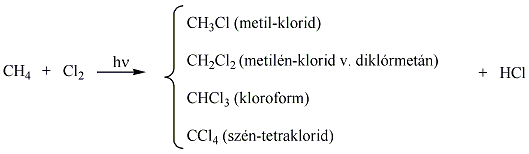
A metán égése erősen exoterm reakció, így fűtésre használják. Fontos vegyipari alapanyag is, vízgőzzel való reakciójakor (1000 °C, nikkel katalizátor) keletkezik a szintézisgáz (CO és H2 bármilyen arányú elegye), melyből számos fontos szerves vegyületet pl.: metanolt vagy formaldehidet állíthatnak elő.
A propán a butánnal együtt a háztartási fűtőeszközök leggyakoribb tüzelőanyaga. A gázüzemű gépjárművekben üzemanyagként használják, az autógáz (LPG) is 95%-ban propán-bután keverékből áll.
A motorbenzinekben a 2,2,4-trimetilpentán (izooktán) rendelkezik a legjobb kompressziótűréssel és az egyenes szénláncú heptán a legrosszabbal. A benzin oktánszámát a vele azonos kompresszótűrésű izzoktán-heptán elegy százalékos összetételével adják meg.
1., Rajzolja fel a következő vegyületek szerkezeti képleteit:
a., 2-metilheptán
b., 4-etil-2-metilhexán
c., 4-etil-3,4-dimetiloktán
d., 2,4,4-trimetilheptán
e., 1,1-dimetilciklopentán
f., 4-izopropil-3-metilheptán
2., Nevezze el az alábbi vegyületeket az IUPAC szabályai szerint!
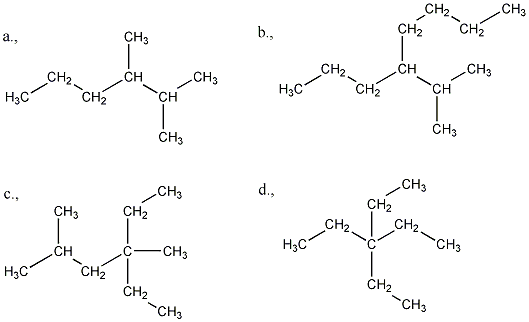
3., Rajzolja fel a C7H16 összegképletű vegyület mind a 9 szerkezeti izomerjét!
4., A következő elnevezések hibásak. Adja meg a vegyületek helyes, az IUPAC szabályoknak megfelelő nevét!
a., 2,2-dimetil-6-etilheptán
b., 4-etil-5,5-dimetilpentán
c., 3-etil-4,4-dimetilhexán
d., 5,5,6-trimetiloktán
5., Egy adott üzemanyag keveréket 87-es oktánszámmal lehet jellemezni. Adja meg az izooktán és a heptán %-os arányát!
6., Rajzolja fel a cisz-1,2-dimetilciklohexán legstabilisabb szék konformációját! A metilcsoportok axiális vagy ekvatoriális térállásúak?
7., Ciklopropánt először 1,3-dibrómpropán és nátrium reakciójával állítottak elő.
a., Írja fel a reakciót!
b., Milyen termék keletkezik az alábbi reakcióban? Milyen lesz a molekula geometriája?
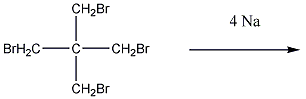
Tartalom
Az alkénekben egy vagy több szén-szén kettős kötés található.
Kis mennyiségben a kőolaj, a földgáz és a kőszénkátrány alkotói. A növényvilágban általánosan elterjedtek, mégpedig főként az izoprén vázas vegyületek körében.
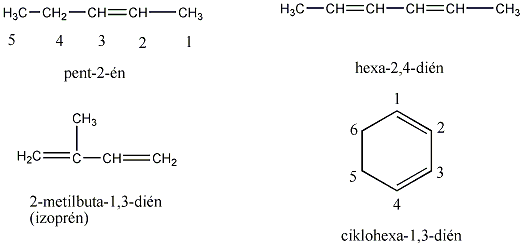
Az alkének és cikloalkének nevét az azonos szénatomszámú alkán és cikloalkán nevéből vezetjük le úgy, hogy az –án végződést –én végződésre cseréljük. A szénláncot úgy számozzuk, hogy a kettős kötés a lehető legkisebb helyzetszámot kapja. Több kettős kötés jelzésére a –dién, -trién, stb. végződések szolgálnak.
A kettős kötések egymáshoz viszonyított helyzete alapján a diolefin izolált, konjugált, vagy kumulált lehet:
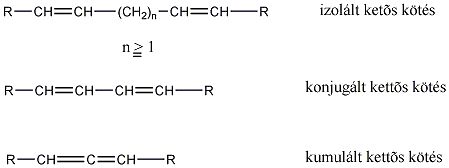
A kettős kötésben résztvevő szénatomok sp2 hibridállapotúak, ennek megfelelően a három hibridpálya sík-trigonális elrendezésű, azaz a kötések egymással 120°-os szöget zárnak be és a kapcsolódó atomok egy síkban találhatók, a pz-pálya pedig erre a síkra merőleges:

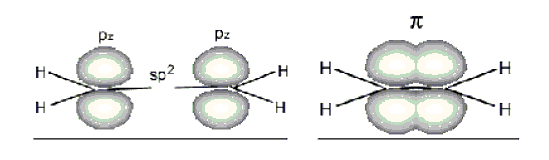
A kettős kötés kötéstávolsága 134 pm, ami jóval rövidebb, mint az etánra jellemző 154 pm C-C kötéstávolság.
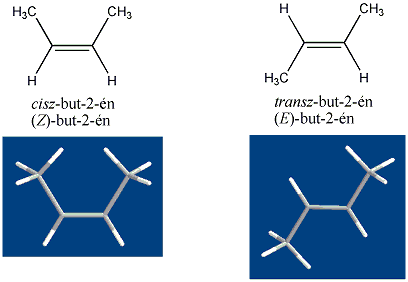
A szén-szén kettős kötés mentén az olefinekben gátolt a rotáció, mert ez a p-pályák átlapolásának a megszűnésével, a -kötés felbomlásával járna. Ezért az 1,2-diszubsztituált etének esetében cisz-transz (Z-E) izoméria jelensége lép fel. Az izomerek közül azt nevezzük cisz-nek, vagy Z-nek (a német zusammen = együtt), amelyikben a két kiválasztott ligandum a sík azonos oldalán helyezkedik el, és transz-nak, vagy E-nek (entgegen = ellentétes) amelyben az ellentétes oldalon találhatók:
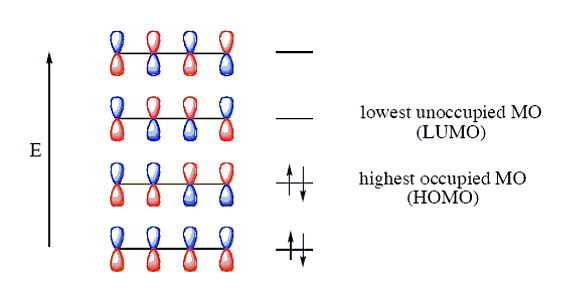
A legegyszerűbb konjugált dién a buta-1,3-dién, melyben valamennyi szénatom sp2 hibridállapotú. A molekula pz-pályái nem csak a -kötések mentén, hanem a -kötés felé is átlapolhatnak, így a négy pz-pályán lévő elektron két delokalizált molekulapályát hoz létre. A butadiénben a C-C egyes kötés rövidebb (0,147 nm), mint az alkánokban (0,154 nm), a kettős kötések viszont hosszabbak (0,136 nm), mint az etén C=C kötése (0,134 nm).
A delokalizáció hatására a vegyület energiatartalma alacsonyabb, mintha két izolált kettős kötést tartalmazna, ezt az energiakülönbséget delokalizációs energiának nevezzük. Az alábbi ábra a buta-1,3-dién molekulapályáit mutatja:
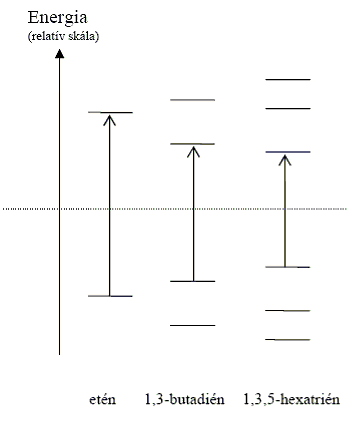
A konjugált kettős kötések számának növekedésével a HOMO és LUMO molekulapályák energiaszintje egyre közelebb kerül egymáshoz, ezért a konjugált poliéneket már a látható fény is gerjeszti, így ezek a vegyületek színesek. A természetben számos ilyen konjugált kettős kötést tartalmazó vegyület fordul elő az izoprén vázas vagy a heterociklusos molekulák között. A konjugáció hatása a HOMO-LUMO pályák távolságára:
Megfelelő vízelvonószer (cc. H2SO4, ZnCl2) hatására, magas hőmérsékleten vízkilépéssel olefinek képződnek:
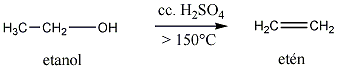
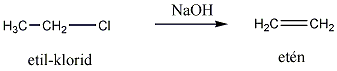
Olvadás és forráspontjuk a szénatomszám növekedésével általában növekszik. A konstitúciós izomereknél az alkánokhoz hasonlóan jelentős eltérések adódhatnak, mivel a kristályszerkezet és a van der Waals-kölcsönhatások is befolyásolják az olvadás- és forráspontot. A cisz és transz izomerek fizikai tulajdonságai is eltérnek egymástól.
Az olefinek jellemző reakciója az addíció. Az addíciós reakció során az olefin kettős kötése felnyílik, a reakciópartner a kettős kötés szénatomjaihoz kapcsolódik, így az sp2 hibridállapotú szénatomok sp3 hibridállapotúvá alakulnak.
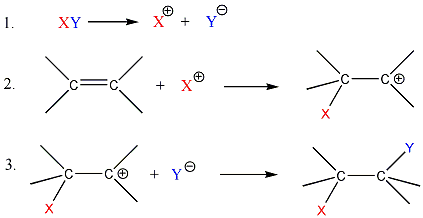
Az elektrofil addíció az alábbi általános reakciólépéseken keresztül játszódik le:
1., Az XY reagens heterolitikusan disszociál, ennek során X+-kation és Y--anion keletkezik.
2., A kation elektrofil támadást indít az olefin elektronban gazdag -kötésére, ekkor karbokation képződik
3., A reakció befejező lépésében a karbokation az Y--anionnal reagál, és így telített vegyület keletkezik.
A következőkben néhány konkrét példa alkének elektrofil addíciós reakcióira:
Az alkének könnyen addícionálnak hidrogén-halogenideket, és alkil-halogenidekké alakulnak. A fenti általános mechanizmus alapján a reakció első lépésében az olefin a protonnal reagál. Ezt követően kapcsolódik a karbokationhoz a halogenidion és az alkil-halogeniddé alakul.
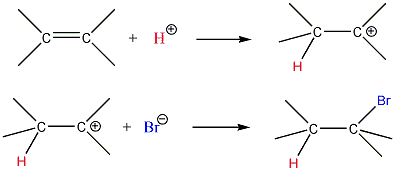
A nem szimmetrikus olefinek, mint pl. a propén hidrogén-bromid addíciójánál kétféle alkil-halogenid képződhet. A főtermék a 2-brómpropán, mellette kis mennyiségben 1-brómpropán is keletkezik. A reakció első lépésében a proton a primer és a szekunder szénhez is kapcsolódhat, azonban a szekunder karbokation képződésének nagyobb a reakciósebessége. A karbokationok relatív stabilitása (tercier>szekunder>primer) meghatározó a termékszerkezet kialakulásában.
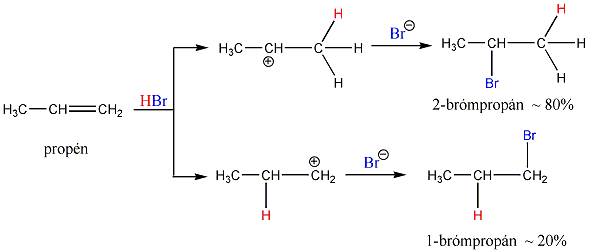
Markovnyikov orosz kémikus már az 1870-es években megfigyelte, hogy a várható két termék közül csak az egyik regioizomer képződik és a következő orientációs szabályt fogalmazta meg: „ az aszimmetrikus olefinek HX-addíciója során a hidrogén mindig ahhoz a szénatomhoz kapcsolódik, amelyik eredetileg is több hidrogént tartalmazott”.
A reakció gyakorlati jelentősége az alkoholszintézis. Savas közegben a Markovnyikov-szabálynak megfelelően játszódik le, az előzőhöz hasonló reakciólépéseken keresztül:
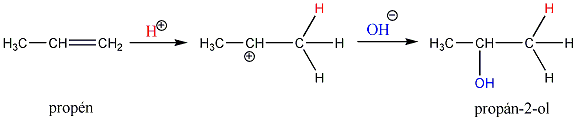
A reakciót a gyakorlatban dihalogén-származékok szintézisére és a kettős kötés kimutatására alkalmazzák – a brómos víz vagy a széntetrakloridos brómoldat elszíntelenedik a reakció során.
Itt a brómmolekulát az olefin nagy elektronsűrűségű -kötése polarizálja, -komplex keletkezik. A reakció következő szakaszában a brómmolekula heterolitikus bomlásával keletkező elektrofil bróm-kation kapcsolódik a kettős kötés mindkét szénatomjához. Az így létrejövő bromóniumiont -komplexnek is nevezik. A reakció utolsó lépésében a nukleofil bromidion a bromóniumion egyik szénatomjához kapcsolódik és így 1,2-dibrómvegyület keletkezik. A sztereokémiát tekintve transz-addíció játszódik le.
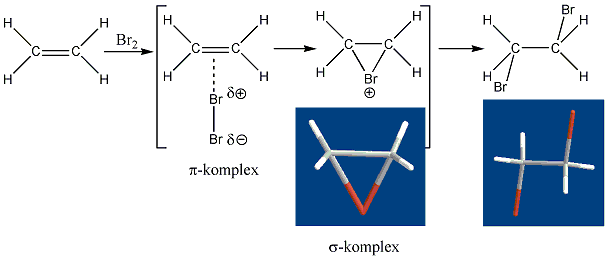
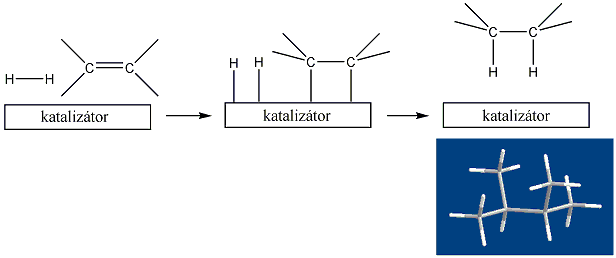
Az alkének az alkalmazott oxidálószertől és reakciókörülményektől függően többféleképpen is átalakulhatnak.
Enyhe körülmények között (hideg KMnO4 hatására) az oxidáció során vicinális diolok képződnek. A reakció lejátszódását a permanganát színének eltűnése jelzi, így ez a reakció is alkalmazható kettős kötés kimutatására (Baeyer-próba):
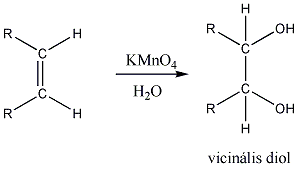
Erélyes oxidáció során (meleg KMnO4) lánchasadás is bekövetkezik és ketonok vagy karbonsavak keletkezhetnek:
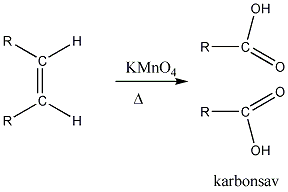
A két német kémikus, Diels és Alder nevét viselő cikloaddíciós reakció egy konjugált dién és egy alkén között játszódik le, gyűrűképződéssel:

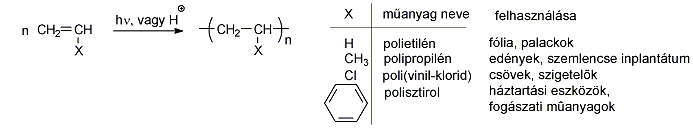
Etén: Az etén a szerves vegyipar legnagyobb mennyiségben gyártott terméke. Kőolajból és földgázból állítják elő. A polimerizációs műanyagok mellett számos más terméket, így etanolt, vinil-acetátot, acetaldehidet, ecetsavat is előállítanak belőle.
Buta-1,3-dién: A szintetikus gumi előállításának alapanyaga. Főleg kőolajból állítják elő.
Sztirol: A polisztirol az egyik legfontosabb műanyag, jó villamos szigetelő. Habosítva hőszigetelő anyagként használják nagy mennyiségben különböző márkaneveken (Hungarocel).
A terpének izoprén – 2-metilbuta-1,3-dién – egységekből felépülő, többnyire növényekben található vegyületek.
Az öt szénatomos izoprénváznak a metilcsoport melletti részét fej (F), a távolabbit láb (L) jelöléssel különböztetjük meg. A terpének szerkezetfelderítése során Ruzicka fogalmazta meg az izoprénszabályt, mely szerint a terpénekben az izoprénegységek szabályos fej-láb illeszkedéssel kapcsolódnak össze. Bár a terpének izoprénegységekből épülnek fel, bioszintézisük során az építőkövük az izopentenil-pirofoszfát illetve annak izomerje, a dimetil-allil-pirofoszfát.
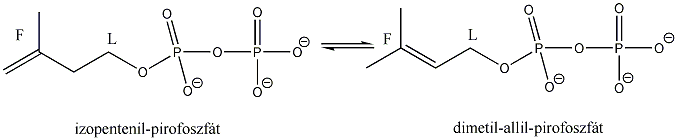
A terpének csoportosítása az őket felépítő izoprénegységek száma szerint történik.
A monoterpének növények illóolajainak fő alkotórészei. Két izoprén egységből, úgynevezett fej-láb, vagy láb-láb kapcsolódással jönnek létre. A monoterpének oxigéntartalmú származékai közül a geraniol és a citrál-a elterjedtek a növényvilágban, az illatszeripar fontos alapanyagai. A monociklusos monoterpének közé tartozik a mentol. Biciklusos monoterpének például a tuja illóolajában található szabinén, a tűlevelűek illóolajában fellelhető - és -pinén és a kámfor.
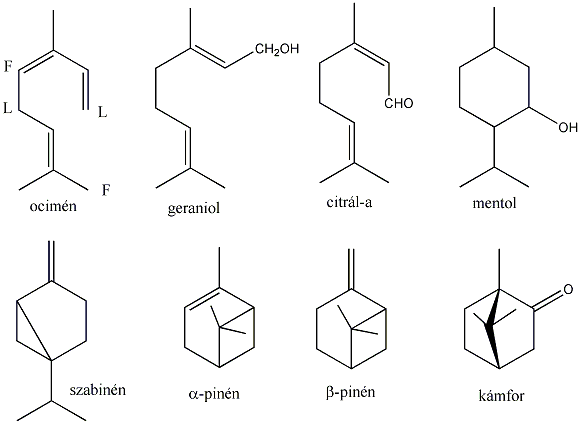
Négy izoprén egységből felépülő vegyületek. Kiemelkedő jelentőségű a klorofill hidrolízisével előállítható fitol.
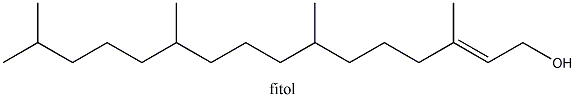
Az A-vitamin vagy retinol a látásban játszik fontos szerepet. A molekula először 11-transz-retinallá oxidálódik, majd a 11-12 kettőskötés izomerizációjával 11-cisz-retinallá alakul.
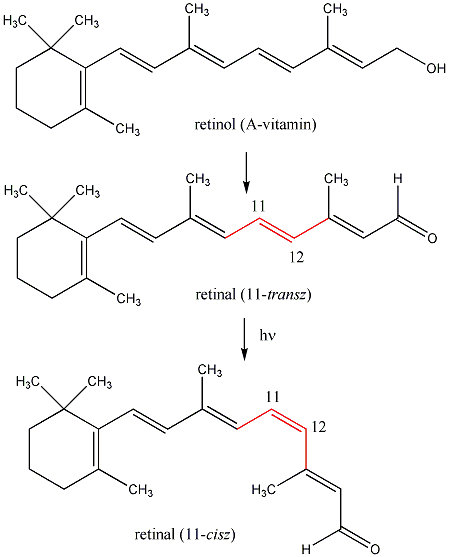
A 11-cisz-retianl az opszin fehérje lizin aminosavjának amino-oldalláncával képez Schiff-bázist. Ez a fényre érzékeny pigment a rodopszin. Fény hatására 11-transz-retinállá izomerizál, így már nem illeszkedik az opszin aktív helyéhez és leválik onnan, az opszin felszabadul. Az izomerizáció konformációs változást okoz az opszin fehérjében, ami a látóidegben kiváltja az ingerületet.
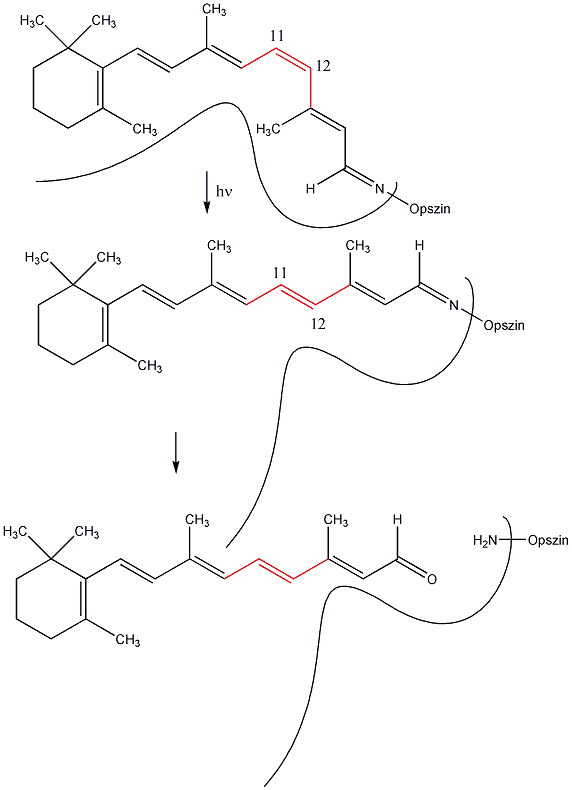
A triterpének hat izoprén egységből levezethető vegyületek. A szkvalén a csukamájolajban, búzacsíraolajban és az élesztőben előforduló vegyület. A koleszterin és más szteránvázas vegyületek bioszintézisének is kulcsintermedierje.
A szteránvázas vegyületek elterjedtek a növény és állatvilágban, kémiai szerkezetük alapját a négy gyűrű kondenzációjával kialakult szterán- vagy más néven gonánváz alkotja.
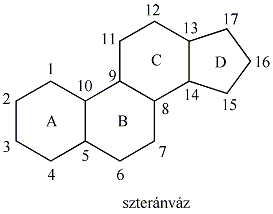
A szteránvázas vegyületeket leggyakrabban biológiai funkciójuk szerint csoportosítjuk.
A koleszterin egy részét a táplálékkal vesszük fel, nagyobb része a májban szintetizálódik. A vérben proteinekhez kapcsolódva, észterként szállítódik a sejtekhez, ott a sejtmembrán felépítésében vesz részt.
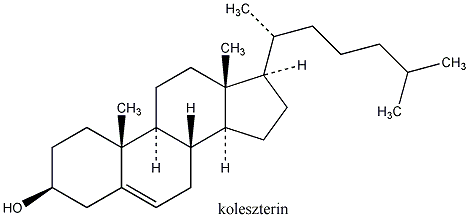
A koleszterin előanyaga számos biológiailag fontos vegyületcsoportnak. A belőle képződő ergoszterinben UV fény hatására a B gyűrű felnyílik, majd kémiai átalakulásokat követően D2-vitamin képződik. A D-vitaminok elsősorban a szervezet kalcium és foszfátanyagcseréjét szabályozzák. Hiányuk következtében zavar áll be a csontok és a fogak fejlődésében. Túladagolása csonttörékenységet okozhat.
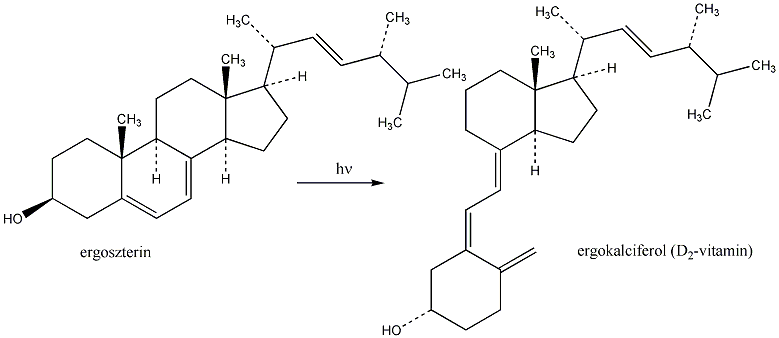
A koleszterolból legnagyobb mennyiségben epesavak képződnek a szervezetben, mint pl. a kólsav. A kólsavmolekula A/B gyűrűjének kapcsolata cisz. A hajlított szerkezet egyik oldalán helyezkednek el az apoláros metilcsoportok és hidrogénatomok, a másik oldalon a karboxil- és hidroxilcsoportok. A fenti szerkezetnek köszönhetően az epesavak és sóik felületaktív vegyületek. Szerepük a zsírok emulgeálása, ezáltal a zsírok emésztésének elősegítése. A víz-lipid határfelületen úgy helyezkednek el, hogy a molekula poláros (OH, COO-) csoportjai a vizes fázis felé, az apoláros csoportok a lipid fázis felé orientálódnak.
Ezek a vegyületek is a koleszterolból keletkeznek, a kezdő szintézislépések a mellékvesekéreg mitokondriumaiban játszódnak le.
A szénhidrát anyagcserét befolyásoló glikokortikoidok jellemző képviselője a kortizon.
A mineralokortikoidok a szervezet sóháztartását befolyásolják. Legjelentősebb képviselőjük az aldoszteron:
A nemi hormonok közül a hím ivari hormonok, az androgének elsősorban a herében, valamint csekély mértékben a mellékvesekéregben is szintetizálódnak. Az androgének alakítják ki a hímekre jellemző másodlagos nemi jelleget, de az általános anyagcserét is fokozzák, serkentik a fehérjék felépítését. Fiziológiailag leghatékonyabb a tesztoszteron.
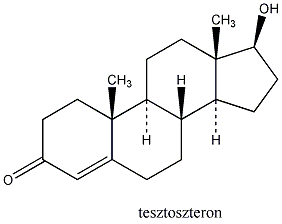
A női nemi hormonok közül az ösztrogének és a progesztagének is a petefészekben szintetizálódnak. Az ösztrogének (pl. az ösztradiol) alapvető biológiai hatása a női másodlagos nemi jelleg kialakításában nyilvánul meg. A progeszteron elsődleges funkciója a terhesség fenntartása és új petesejt érésének meggátlása.
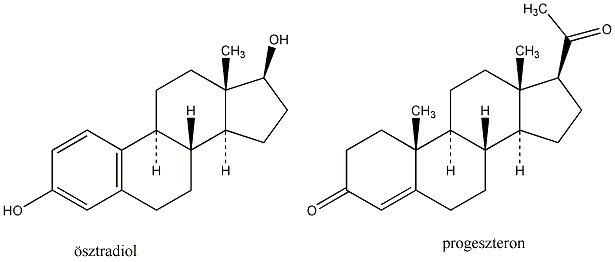
A szteroidglikozidok legfontosabb csoportját a növényi eredetű, szívre ható szteroidok alkotják. Gyógyszerként való felhasználásuk azon alapul, hogy fokozzák a szívizom kontrakcióját. Ezen hatásuk miatt azonban súlyos mérgezést is okozhatnak. Egyik legjelentősebb képviselőjük a digitoxigenin:
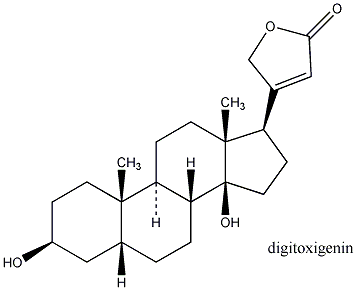
A növényekben található -karotin az A-vitamin provitaminja. Enzimkatalizált kettőskötés hasadással retinol keletkezik belőle. A karotinoidok a lánc végén található gyűrű szerkezetében különböznek egymástól. A -karotinban a lánc mindkét végén -jonon, az -karotinban az egyik végén -, a másikon -jonon, míg a -karotinban az egyik végen -jonon gyűrű van, míg a másik láncvég nem záródott gyűrűvé. A karotinoidok a bennük lévő konjugált kettős kötések miatt színesek, mert a -elektronjaikat már a látható fény is gerjeszti.
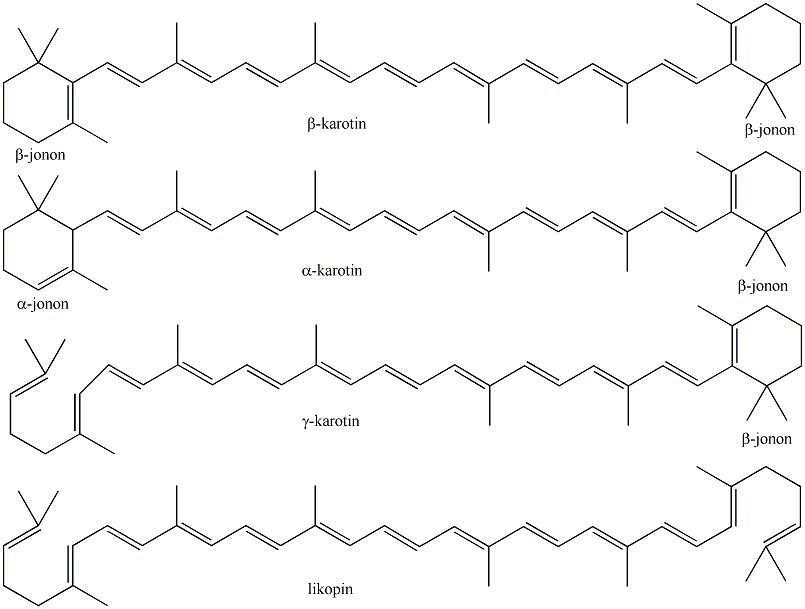
1., Nevezze el az alábbi vegyületeket az IUPAC szabályainak megfelelően!
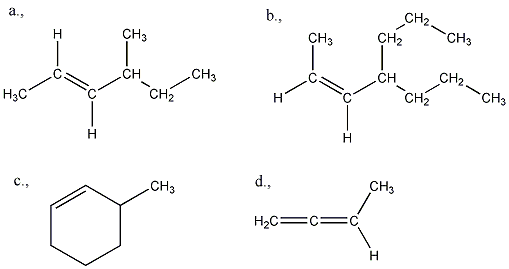
2., Rajzolja fel az alábbi vegyületek szerkezeti képletét!
a., 2-propilhept-2-én
b., 2,4-dimetilhex-2-én
c., okta-1,5-dién
d., 4-metilpenta-1,3-dién
e., cisz-4,4-dimetilhexa-2-én
f., (E)-3-metilhept-3-én
3., Az alábbi molekulák közül melyiknél fordulhat elő cisz-transz izoméria?
4., 2-Metilpropén és sósav reakciójával milyen termék keletkezik? Rajzolja fel a kiindulási és a végtermék szerkezetét!
5., A növényekben a nagyobb izoprénvázas vegyületek az őket felépítő izoprén egységekre bonthatók.
a., A likopin és a -karotin molekulában is jelölje az izoprén egységeket!
b., A növényekben a -karotin likopinból keletkezik. Jelölje a gyűrűzáródás során kialakuló -kötéseket!
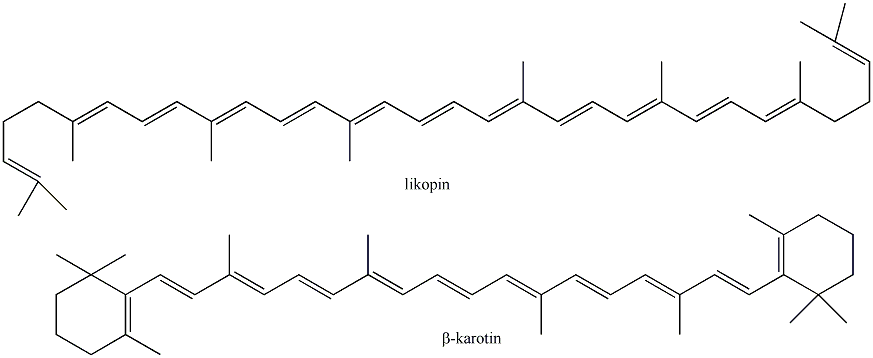
6., Milyen termékek keletkeznek az alábbi reakciókban? Ahol regioszelektív a reakció, ennek eredményét a termékekben is jelölje! (Az aromás gyűrű nem lép reakcióba a felhasznált reagensekkel.)
7., Írja fel a ciklohexén brómmal való reakcióját a köztitermékek és a megfelelő sztereokémia jelölésével!
8., Mi volt a kiindulási alkén szerkezete, ha az alábbi termékek keletkeztek?
a., alkén + Br2 → 2,3-dibróm-5-metilhexán
b., alkén + HBr → 2-bróm-3-metilheptán
Tartalom
Az alkinok nevét az azonos szénatomszámú telített szénhidrogének nevéből vezetjük le úgy, hogy az –án végződést –in végződésre cseréljük. A számozást arról a láncvégről kezdjük, amelyikhez közelebb esik a hármas kötés.
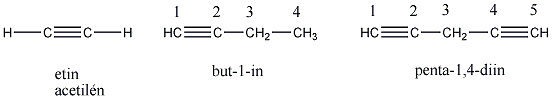
Az acetilénben a szénatomok sp hibridállapotúak. A szénatomok két -kötést létesítenek, melyek lineárisak. A py- és pz-pályák egymásra merőleges -kötéseket alakítanak ki. Az így kialakult hármas kötések hossza jelentősen kisebb, mint a kettős kötésé, az etinben 121 pm.

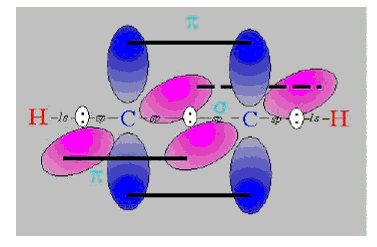
Fizikai tulajdonságaik hasonló módon változnak a szénatomszám függvényében, mint az alkánok és alkének fizikai tulajdonságai.
Az acetilénszármazékok a két -kötés miatt reakcióképes, nagy energiájú vegyületek. Az olefinekhez hasonlóan addícióra hajlamosak, emellett azonban az sp-hibridállapotú szénatomhoz kapcsolódó hidrogénatom gyengén savas jellegű.
A sósavvaddíció higany(II)-klorid katalizátor felületén játszódik le. Először szubsztituált olefin képződik, majd tovább reagálhat újabb HCl molekulával. A reakció iparilag nagy jelentőségű, mert az első lépésben keletkező vinil-klorid a PVC monomerje.
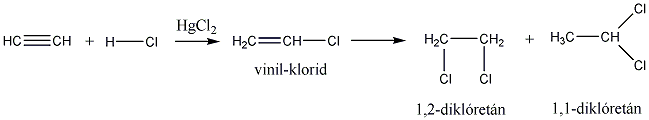
Az acetilén vízaddíciójával acetaldehid keletkezik. A reakcióban először vinil-alkohol képződik, ami keto-enol tautomerizációval azonnal acetaldehiddé alakul:
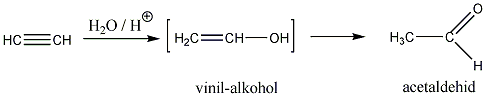
Az acetilén hevesen reagál halogénekkel, két mól bróm addíciójával tetrabróm-etán képződik:
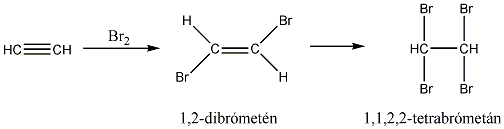
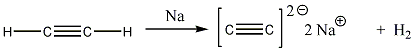
Acetilén: nagy energiatartalmú, instabil vegyület. Ipari jelentősége nagy, mert a vinil-kloridhoz hasonlóan más származékai (vinil-éterek, vinil-észterek) is fontos műanyag alapanyagok. Nagy égéshője miatt hegesztéshez használják. Az oxigénnel kevert acetilénláng hőmérséklete eléri a 3000 °C-t is.
1., Írja fel az alábbi vegyületek szerkezeti képletét!
a., 3-etilhept-1-in
b., 3,5-dimetilhex-4-én-1-in
c., hepta-1,5-diin
d., 1-metilciklopenta-1,3-dién
2., Milyen termékek keletkeznek az alábbi reakciókban?
a., hex-1-in + 1 ekvivalens HBr →
b., hex-1-in + 1 ekvivalens Cl2 →
c., hex-1-in + H2, Lindlar katalizátor →
3., Rajzolja fel a kiindulási alkinok szerkezetét, amelyekből vízzel való reakciójuk során az alábbi ketonok keletkeznek:
4., But-1-in-ből kiindulva milyen reagensek felhasználásával lehet az alábbi termékekekt előállítani (esetleg többlépéses reakcióban)?
a., bután
b., 1,1,2,2-tetraklórbután
c., 2-brómbután
d., bután-2-on
Tartalom
Az aromás vegyületek olyan gyűrűs molekulák, amelyek nagyfokú telítetlenségük ellenére a telített vegyületekre jellemző tulajdonságokat mutatnak.
A legegyszerűbb aromás szénhidrogén a benzol. 1825-ben Michael Faraday fedezte fel a kőszénkátrányban és hamarosan megállapították, hogy az összegképlete C6H6. A benzolmolekulában a szén és hidrogén aránya 1:1, akárcsak az acetilénben, ami a molekula telítetlen jellegére utal. Ennek ellenére a benzol reakciókészsége eltér a telítetlen vegyületekétől. Így például hidrogénezése csak lassan és nehezen játszódik le, nem az addíciós, hanem a szubsztitúciós reakciók a jellemzők.
Az aromás szénhidrogének a benzolhoz hasonlóan nagyobb mennyiségben a kőszénkátrányból nyerhetők, de számos képviselőjük növényi illóolajokban, gyantákban, balzsamokban is előfordul.
Az aromás szénhidrogének monociklusos származékait általában a benzol szubsztituált származékaiként nevezzük el, azonban néhány rövidebb alifás oldalláncot tartalmazó származéknak triviális neve is van:
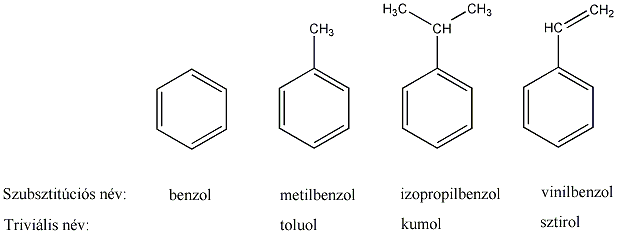
Ha a benzolmolekula két hidrogénatom helyett valamilyen szubsztituenst tartalmaz, három helyzeti izomert kapunk, függetlenül attól, hogy a szubsztituensek azonosak-e vagy sem. A szubsztituensek helyzetét számokkal, vagy o- (orto), m- (meta), p- (para) előtaggal jelölhetjük:
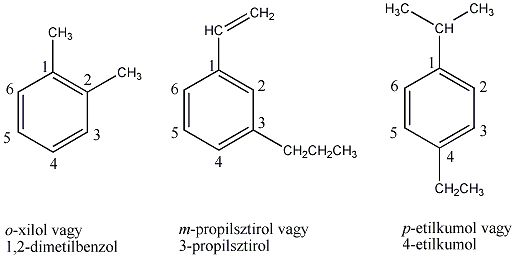
A benzolmolekulában a szénatomok sp2 hibridállapotúak, ezért sík trigonális szerkezetűek. A gyűrűt alkotó hat szénatom és a hozzájuk kapcsolódó hidrogénatomok egy síkban helyezkednek el, a szénatomok szabályos hatszöget alkotnak. A szén-szén kötéshossz értéke 0,139 nm, azaz rövidebb, mint a szén-szén egyes kötéseké (0,154 nm), de hosszabb, mint a kettős kötés (0,134 nm) értéke. Minden szén-szén kötés hossza azonos, átmenet az egyes és kettős kötés között. Az sp2 hibridállapotú szénatomok p-elektronjai olyan molekulapályákon helyezkednek el, amelyek a gyűrű síkja alatt és felett egyenletesen oszlanak el, delokalizálódnak. A benzolmoleklulában gyűrűs konjugált elektronrendszer, ún. -elektronszextett alakul ki.
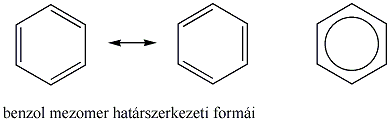
Aromás rendszerről akkor beszélhetünk, ha az alábbi három feltétel teljesül:
- a gyűrűt alkotó atomok egy síkban helyezkedjenek el – a gyűrű koplanáris legyen
- valamennyi gyűrűt alkotó atom rendelkezzen pz-atompályával
- a delokalizációban 4n+2 számú (n = 0, vagy pozitív egész szám) p-elektron vegyen részt – Hückel-szabály
A benzol és homológjai nagy mennyiségben a kőszénkátrányban és különböző kőolajféleségekben fordulnak elő, melyekből frakcionált desztillációval nyerhetők ki. A cikloparaffintartalmú ásványolaj frakciók dehidrogénezésével is aromás vegyületekhez juthatunk:
A monociklusos aromás szénhidrogének színtelen, szobahőmérsékleten általában folyékony, jellegzetes illatú anyagok. Vízben nem, de szerves oldószerekben jól oldódnak.
Az aromás vegyületeket az aromás kötésrendszer stabilitása jellemzi. Az aromás szénhidrogének addíciós reakciókban nehezen, szubsztitúciós reakciókban szívesebben vesznek részt.
Benzol és bróm elegye vaskatalizátor jelenlétében enyhe melegítés hatására brómbenzollá alakul, aromás elektrofil szubsztitúciós reakció játszódik le.
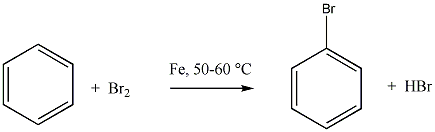
Az aromás gyűrű -elektronjai nukleofil jellegűek, míg a reaktáns elektrofil. A reakció több lépésen keresztül játszódik le, ezek a következők:
a., A benzol és a brómmolekula kölcsönösen polarizálják egymást, a kölcsönhatás eredményeképpen mindkét molekulában torzul az elektroneloszlás, dipólussá válnak.
b., A katalizátor elősegíti a reaktáns molekula heterolitikus hasadását, azaz a brómmolekulából bromidion és brómkation képződik.
c., A brómkation és a benzol -elektronrendszere között -komplex képződik.
d., Az aromás elektronrendszer átmenetileg megszűnik, mert két -elektron és a brómkation -kötést alakít ki, -komplex jön létre. A gyűrűben maradt 4 -elektron és a kialakult pozitív töltés öt szénatomon delokalizálódik.
e., Ezután a -komplex elbomlik, egy proton lehasadásával visszaáll az aromás -elektronszextett. A -komplex elbomlását az FeBr4- anion segíti elő.
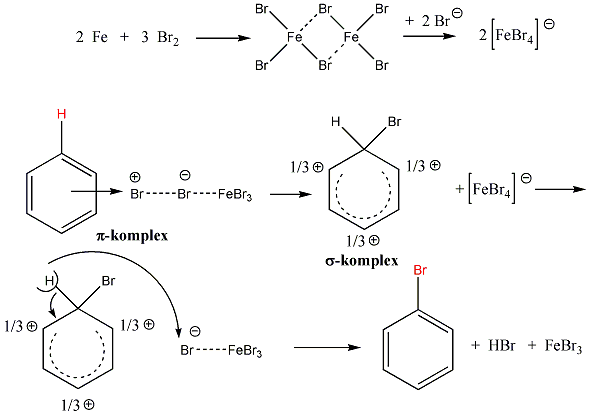
A benzol nitrálása kénsav és salétromsav 2:1 arányú elegyében könnyen lejátszódik, a brómozáshoz hasonló mechanizmussal. A két sav közül a kénsav az erősebb sav, ezért az első lépésben protonált salétromsav keletkezik, majd a második lépésben egy újabb kénsavmolekula alakítja ki az elektrofil nitrónium kationt:
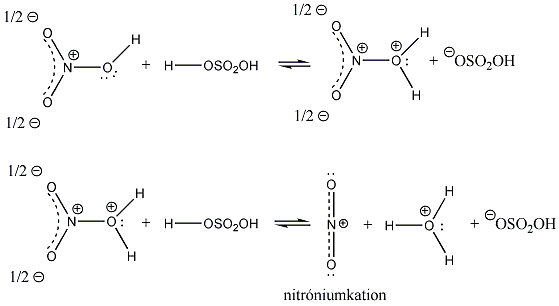
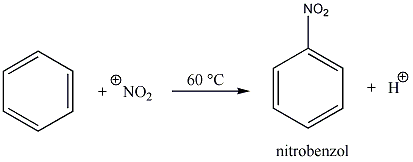
Az aromás szénhidrogének alkil-halogenidekkel Lewis-sav, olefinekkel pedig erős sav jelenlétében alkilezhetők. A Lewis-sav hatására az alkil-halogenidből először primer karbéniumion keletkezik, ami könnyen átalakul a stabilisabb szekunder karbéniumionná. Az olefinek erős savak (HBr, H2SO4) hatására protonálódnak, és ekkor szintén karbéniumion keletkezik:
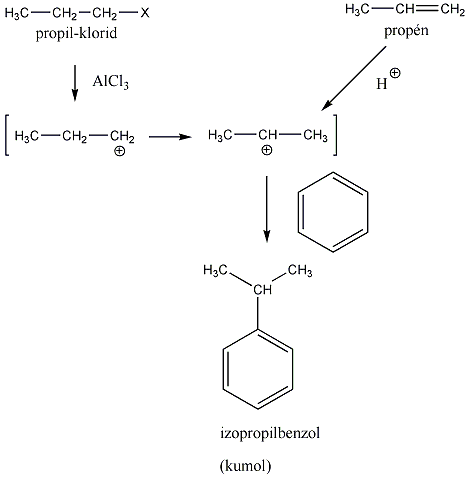
Az egyszeresen szubsztituált aromás gyűrűn a második szubsztituens belépésére több lehetőség is kínálkozik: statisztikailag az várnánk, hogy 40% orto-szubsztituált, 40% meta-szubsztituált és 20% para-szubsztituált termék keletkezik.
A brómbenzol további brómozásakor valójában azonban csak para-dibrómbenzol keletkezik. A nitrobenzol nitrálásakor pedig csak meta-helyzetbe lép be a következő nitro-szubsztituens:
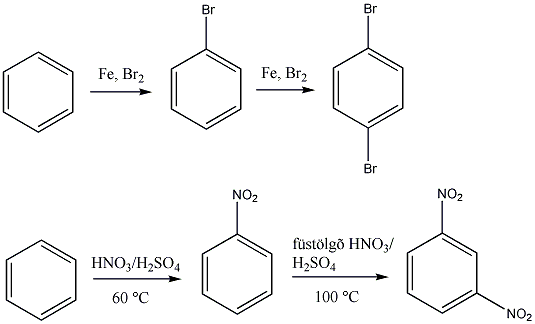
Tehát, a fenti reakcióegyenletek és reakciókörülmények azt mutatják, hogy:
- az elektrofil szubsztitúció regioszelektíven játszódik le
- a második nitrocsoport beviteléhez erélyesebb körülmények kellenek
I. osztályú csoportok: orto- és para-helyzetbe irányítanak
- megkönnyítik a reakciót: pl.: -CH3, -OH, -OR, -NH2, -NR2
- megnehezítik a reakciót: pl.: -Hlg
II. osztályú csoportok: meta-helyzetbe irányítanak és megnehezítik a második
szubsztituens belépését (-NO2, -CHO, -COR, -CN, -COOH)
A s-komplex stabilitását befolyásoló tényezők: a kationban a pozitív töltést a szénatomok viselik, de ez nem egyenletes, hanem a konjugált láncolat páratlan tagjain oszlik meg.
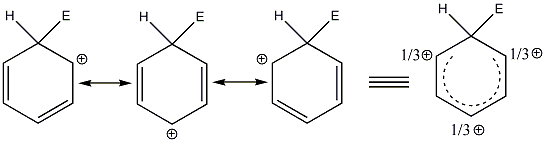
Az alábbi ábrákon jelölt töltéseloszlás jól szemlélteti, hogy az egymáshoz képest orto- vagy para-helyzetben lévő szubsztituensek olyan szénatomhoz kapcsolódnak, amelyik részleges pozitív töltéssel rendelkezik, ezért elektromos hatásuk jobban befolyásolja a -komplex stabilitását, mint a meta-szubsztituens.
A metilcsoport elektronküldő szubsztituens (+I effektus), ezért csökkenti a kation elektronhiányát. Ez a stabilizáló hatás különösen azokban a s-komplexekben érvényesül, amelyekben az elektrofil reagensek a metilcsoporthoz képest orto- vagy para-helyzetűek:
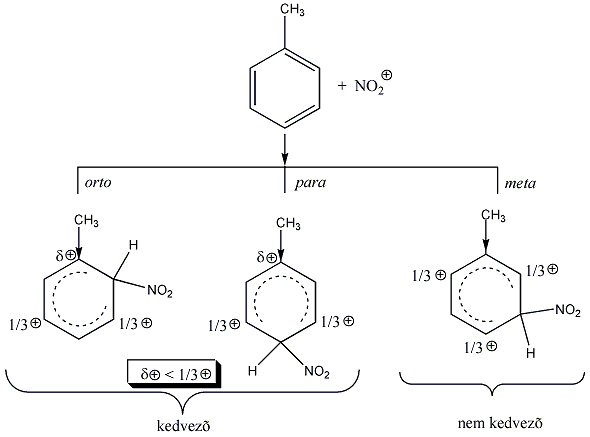
A nitrocsoport erős elektronszívó hatása (-I, -M effektusa) miatt még nagyobb lesz az elektronhiány a s-komplexben. Ez a helyzet különösen kedvezőtlen akkor, ha olyan szénatomhoz kapcsolódik, amelyik a komplexben a pozitív töltést viseli. Kevésbé kedvezőtlen, ha meta-helyzetet foglal el:
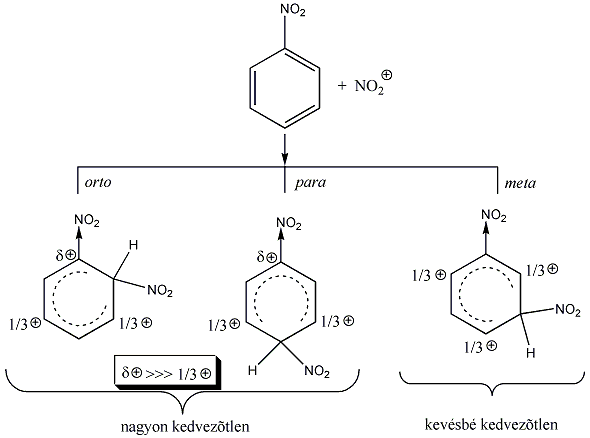
Az elektromos hatás mellett a kapcsolódó szubsztituens térigénye, polarizációja és elektronegativitása is befolyásolja az irányító hatást.
Ha valamilyen heteroatom (pl. N vagy O) kapcsolódik a gyűrűhöz, akkor a heteroatom nemkötő elektronpárja és a gyűrű -elektronjai delokalizálódnak (ld. aromás aminok). Így pl. mivel a C és a N elektronegativitásának különbsége kicsi, a nitrogén nemkötő elektronpárja már alapállapotban is delokalizálódik a gyűrű felé. A kialakuló s-komplexben ez még fokozódik, így csökkenti az elektronhiányt, stabilizálja az átmeneti állapotot.
Ezzel szemben a fluor elektronegativitása lényegesen nagyobb (DEN = 1.5), ezért a delokalizáció mértéke sokkal kisebb lesz. Azonban a fluoratom nemkötő elektronpárjának és az aromás gyűrű -rendszerének kölcsönhatásából (+M) származó aktiváló hatás ellensúlyozza a nagy elektronegativitásbeli különbségből (-I) származó dezaktiváló hatást. Ennek köszönhetően a halogénatomok ugyan orto- és para-helyzetbe irányítanak, de megnehezítik a következő szubsztituens belépését, dezaktiváló hatásúak.
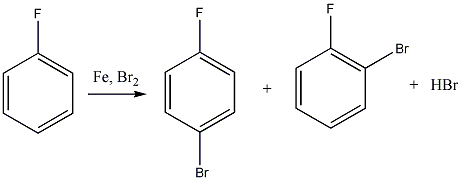
A különböző szubsztituensek irányító és reakciósebességre gyakorolt hatását a következő táblázat foglalja össze:
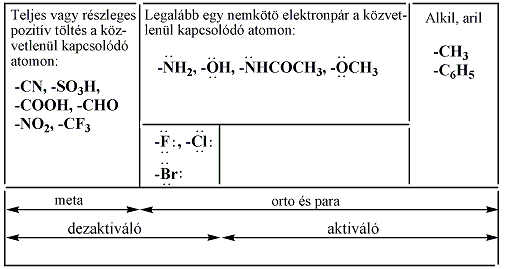
Az oldalláncot tartalmazó aromás szénhidrogének UV fény hatására gyökös mechanizmusú reakcióban reagálnak brómmal, így a toluolból benzil-bromid keletkezik:
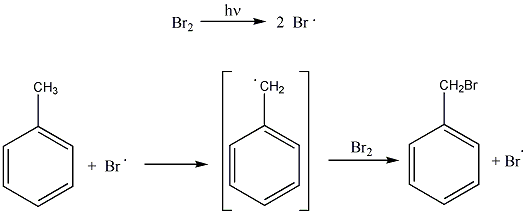
Az oldalláncban való brómozáshoz hasonlóan az aromás gyűrűhöz kapcsolódó lánc oxidációja is gyökös intermedieren keresztül játszódik le az oxidálószer (KMnO4, CrO3) hatására. Az alkil oldallánc mindig az -szénatomon oxidálódik, így pl. az etil-benzolból és a toluolból is benzoesav keletkezik:
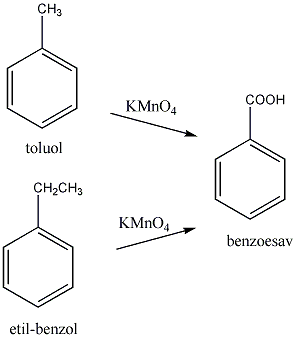
A benzol erősen mérgező, rákkeltő hatású vegyület. Belélegzése eszméletvesztést, nagyobb mennyisége halált okoz. Nagy mennyiségben használja fel a vegyipar oldószerek, gyógyszerek, festékek, robbanóanyagok és növényvédő szerek előállítására.
A toluolt festékek, lakkok oldószereként alkalmazzák, a 2,4,6-trinitrotoluol gyártásának egyik alapanyaga. Szerves szintézisek kiindulóanyagaként hasznosítják.
Kondenzált policiklusos aromás szénhidrogének: naftalin, antracén, fenantrén
Mindhárom vegyület a kőszénkátrány ismert alkotója.
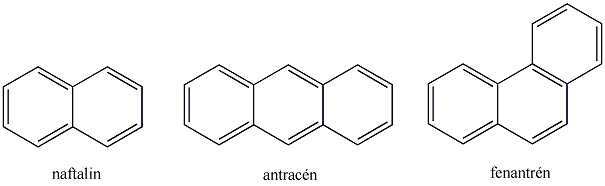
1., Írja fel a következő elnevezéseknek megfelelő szerkezeteket!
a., m-brómfenol
b., 1,3,5-trihidroxi-benzol
c., p-jód-nitrobenzol
d., 2,4,6-trinitrotoluol
e., o-amino-benzoesav
f., 3-metil-2-fenilhexán
2., Rajzolja fel az alábbi vegyületek minden lehetséges izomerét!
a., dinitrobenzol
b., bróm-dimetilbenzol
3., Milyen termékek keletkeznek, ha az alábbi vegyületeket nitráljuk (csak egy nitrocsoport beépülésével számolunk)?
a., brómbenzol
b., benzonitril
c., benzoesav
d., nitrobenzol
e., fenol
f., benzaldehid
4., Állítsa sorrendbe az alábbi vegyületeket az aromás elektrofil szubsztitúcióban mutatott reakciókészségük alapján! A legreakcióképesebbel kezdje a sort!
benzol, nitrobenzol, fenol
5., Az alábbi aromás vegyületben a trimetilammónium csoport aktiváló vagy dezaktiváló csoportként viselkedik aromás elektrofil reakcióban? Indokolja a választ!
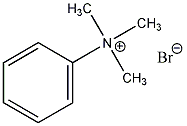
Ezek a vegyületek különböző szénhidrogének egy vagy több hidrogénatomjának halogénatomra történő kicserélésével vezethetők le. A vegyületcsoportnak a tagjai általában jelentős reakciókészséggel rendelkeznek, így sok laboratóriumi és ipari szintézisnél használatosak.
Csoportosítás
A halogénezett vegyületeket többféle szempont szerint csoportosíthatjuk:
-
a halogénatomok minősége (fluorozott, klórozott, brómozott és jódozott származékok),
-
a halogénatomok száma (mono-, di-, tri-, polihalogén-származékok),
-
több halogénatomot tartalmazó származékok esetében a halogénatomok viszonylagos távolsága (geminális, vicinális, diszjunkt)
-
a halogénatom(ok)hoz kapcsolódó szénatom rendűsége
-
a halogénatom(ok)hoz kapcsolódó szénváz minősége (telített, telítetlen, aromás),
-
a halogénatom és a hozzá kapcsolódó szénváz jellemző szerkezeti részlete (pl. kettőskötés, vagy aromás gyűrű, így az α,ß-helyzetű olefinkötést tartalmazó származékot vinil-halogenidek nevezzük)
Példák különböző minőségű és számú halogénatomokat tartalmazó származékokra:
 |
 |
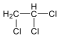 |
|
monobróm-származék |
diklór-származék |
triklór-származék |
Példák több halogénatomot tartalmazó vegyületekben a halogénatomok viszonylagos távolságára:
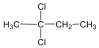 |
 |
 |
|
geminális dihalogén vegyület |
vicinális dihalogén vegyület |
diszjunkt dihalogén vegyület |
Példák a halogénatomhoz kapcsolódó szénváz minőségére, a halogénatomhoz kapcsolódó különböző rendű szénatomokat tartalmazó vegyületekre.
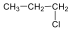 |
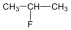 |
 |
|
alkil-halogenid (primer) |
alkil-halogenid (szekunder) |
alkil-halogenid (tercier) |
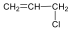 |
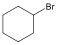 |
 |
|
alkenil-halogenid |
cikloalkil-halogenid |
aromás halogenid |
Izomériaviszonyok
A halogénatom(ok) beépülésével az analóg szénhidrogénekhez képest megnő a szerkezeti izomerek száma. Pl. a propánnak, amelynek nincs izomerje, az egy klór belépésével képződő klór-propánnak kettő, kettő beépülő klóratom esetén a diklór-propánnak már 4 szerkezeti izomerje lehetséges.
Nómenklatúra
A halogénezett szénhidrogének szisztematikus elnevezésére a szubsztitúciós és a csoport-funkciós nómenklatúra is alkalmazható.
A szubsztitúciós nómenklatúra értelmében a halogén-származékot annak a szénhidrogénnek származékaként nevezzük el, amelyből a hidrogén/halogén cserével levezethető.
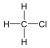 klór-metán |
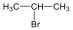 2-bróm-propán |
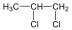 1,2-diklór-propán |
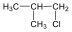 1-klór-2-metilpropán | |
 bróm-benzol |
A halogénezett szénhidrogén számozását az alapszénhidrogén számozása szabja meg, de az alapszénhidrogén számozásával összeegyeztethető legkisebb helyzetszámokkal kell megadnunk. Amennyiben az alapszénhidrogén főláncának kiválasztására vonatkozó szabály megengedi, a főláncot úgy kell kiválasztanunk, hogy a halogénatom(ok) ehhez kapcsolódjon(ak).
A csoport-funkciós nómenklatúra szerint megnevezzük a halogénatomhoz kapcsolódó csoportot majd a halogén nevét, utóbbit -id végződéssel. Több azonos halogénatom esetén a halogének számára utaló előtagot használunk.
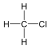 metil-klorid |
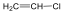 vinil-klorid |
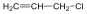 allil-klorid |
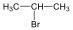 izopropil-klorid | |
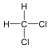 metilén-diklorid |
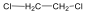 etilén-diklorid |
 benzil-klorid |
 benzilidén-triklorid |
A fentieken túlmenően használatosak még triviális nevek is, pl. kloroform (CHCl3), jodoform (CHI3), stb..
A halogénezett vegyületek előállítása
Szénhidrogénekből kiindulva közvetlen halogénezéssel, halogén-, ill. hidrogén-halogenid-addícióval, vagy speciális szubsztitúciós reakciókkal juthatunk halogénezett vegyületekhez.
A gyakorlatban a szénhidrogének fluorozását, klórozását, brómozását alkalmazzák, jóddal közvetlen módon halogénezést nem lehet végezni. A fluor viszont robbanásszerűen reagál pl. az alkánokkal, lánchasadás is bekövetkezik, ezért csak speciális módon lehet a direkt fluorozást alkalmazni. Emiatt a fluor- és jódszármazékokat más úton állítják elő.
Telített szénhidrogénekből gyökös láncreakcióval képződnek alkil-halogenidek. Ilyen reakció pl. a metán klórozása, vagy brómozása.
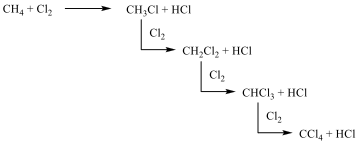
Az elemi klór és bróm esetében UV-fénybesugárzást, vagy termikus aktiválást kell alkalmazni. A reakció több termék képződéséhez vezet. Az egyes termékek arányát a metán-klór arányával és a hőmérséklettel lehet változtatni. Elemi klór helyett gyakran szulfonil-kloridot is használnak. Ha különböző rendű szénatomokat tartalmaz a kiindulási szénhidrogén, akkor különbség figyelhető meg a reakciósebességben. A sorrend: tercier>szekunder>primer.
Ezen az úton cikloalkánokat is halogénezhetünk. Egyedül ciklopropil-származékok előállítása nem valósítható meg, mert a reakció körülményei között felnyílik a gyűrű.
Telítetlen szénhidrogénekre (olefinekre és acetilénekre) könnyen addícionálhatók elemi halogének, ill. hidrogén-halogenidek
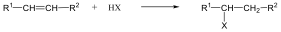
Az addíció a Markovnyikov-szabály szerint játszódik le, amennyiben nincsenek jelen peroxidok. A reakció végeredménye anti-Markovnyikov-szabály szerinti, ha az addíció során peroxidok vannak jelen, mert ebben az esetben gyökös mechanizmussal játszódik le a reakció.
A hidrogén-halogenidek (HX) addíciós készsége a halogén atomtömegének növekedésével nő, a HF addíciójának nincs gyakorlati jelentősége. A szabad halogének addíciós készsége viszont éppen ellentétes módon változik, a halogén méretének növekedésével csökken, így a jód addíciója preparatív szempontból nem alkalmazott.
Az elemi halogének addíciója során orientációs jelenségek nem lépnek fel: olefinekből mindig vicinális, acetilénekből telítetlen vicinális di-, majd 1,1,2,2-tetrahalogén-származékok képződnek.
Az aromás szénhidrogének két úton alakíthatók át halogénezett származékokká, vagy addíciós, vagy szubsztitúciós reakcióval. Megkülönböztetjük a halogénezési reakciót aszerint is, hogy a halogén beépülése az aromás gyűrűt (magot), vagy az oldalláncot érinti.
Lewis-savak jelenlétében, sötétben az aromás gyűrű halogéneződik.
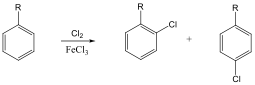
Magasabb hőmérsékleten és UV-fénnyel történő besugárzás hatására az oldalláncban történik szubsztitúció.
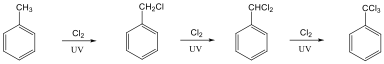
A maghalogénezési reakciót iparilag az ún. oxiklórozásnál alkalmazzák. A reakciót oldatban, nitrátionok jelenlétében hajtják végre.
Benzil-klorid-típusú halogénszármazékokat az ún. klórmetilezési reakcióval lehet előállítani. Az aromás szénhidrogént vízmentes cink-klorid és paraformaldehid jelenlétében száraz sósavgázzal reagáltatják.
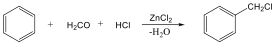
Alkil-halogenidek halogéncserés (SN) reakciójával is eljuthatunk halogénezett származékokhoz. Pl. alkil-kloridot acetonban nátrium-jodiddal melegítve alkil-jodid állítható elő.

A korábban hűtőgépekben és különböző spray-k hajtógázként, ma már ezekre a célokra betiltott freonokat alkil-kloridok hidrogén-fluoriddal végrehajtott halogéncserés reakciójával állították elő, antimon-(III)-, vagy antimon(V)-klorid katalizátor jelenlétében. A következő példa az ún. Freon-22 szintézisét mutatja.
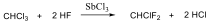
Alkoholokból többféle módon is a megfelelő halogenidekhez juthatunk. Hidrogén-halogenidekkel is átalakíthatjuk az alkoholokat.

Ez egy általában lassú egyensúlyi reakció, ami a reakciókörülmények által meghatározott egyensúly beállásáig játszódik le, de az egyensúly eltolható a kívánt származék irányába a termék folyamatos eltávolításával, vízmegkötéssel, vagy nagy savfelesleg esetén. A hidrogén-halogenidek nem egyforma sebességgel reagálnak: HF<<HCl<HBr<HI. Jódszármazékokat vörösfoszforból és jódból in-situ preparált foszfor-trijodiddal állítanak elő, mert HI alkalmazása esetén szénhidrogének is képződhetnek.

Az alkoholok rendűsége is befolyásolja a reakciósebességet. A sorrend tercier>szekunder>primer szerint változik.
A tercier alkoholok általában szobahőmérsékleten átalakulnak,

a primer alkoholok és sósav reakcióelegyhez a melegítés mellett még vízmegkötő anyagra is, pl. cink-kloridra is szükség van.
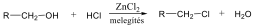
Az alkoholok karbonsavhalogenidekkel és szervetlen halogénezőszerekkel (pl. PBr3, PCl3, PCl5, SOCl2, SF4) az előzőek szerint átalakíthatók.
A telítetlen halogénszármazékok közül a vinil- és allil-halogenidek a legfontosabbak. Előállítási példákat ezekre mutatunk be.
Vinil-klorid acetilénből állítható elő sósav addícióval.
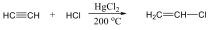
Allil-klorid propén klórozásával állítható elő. A reakciót magas hőmérsékleten (500 C) hajtják végre.
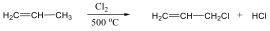
Allilhelyzetű szénatom hidrogénje lecserélhető halogénre az ún. Ziegler-féle módszerrel, amelyben pl. N-bróm-szukcinimidet használnak halogénező szerként.

Előny, hogy alkalmazható a halogéncserés módszer nem csak telített láncokat tartalmazó vegyületeknél, hanem telítetleneknél is, mono-, di és polihalogénszármazékok szintézisére.
Aminokból is van lehetőség halogéntartalmú származékok előállítására. Ennek a módszernek az aril-halogenidek esetében van gyakorlati jelentősége. A primer aromás aminokat először diazotálják, majd a képződő diazóniumsót aril-halogeniddé alakítják. Szükség szerint alkalmazni kell katalizátort, amit főleg a halogén minősége határoz meg.
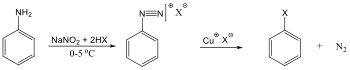
X=Cl,Br
Oxovegyületekből foszfor(V)-halogeniddel (X=Cl, Br), vagy kén-tetrafluoriddal geminális dihalogén származékokat lehet előállítani.
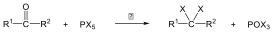
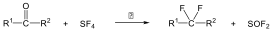
Ha a fenti általános képlettel jellemzett oxovegyületben az egyik R-csoport metilcsoport és lúgos közegben klórral, brómmal vagy jóddal reagáltatják, akkor a metilcsoport átalakul a trihalogén-metánná, haloformmá. Ez a reakció nem csak preparatív szempontból fontos, hanem analitikai, kimutatási módszernek is használható.
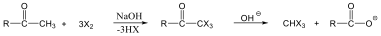
Karbonsavak higany-, vagy ezüstsóiból is állíthatók elő halogéntartalmú szénhidrogének, de a kiindulási vegyületnél egy szénatommal rövidebbek lesznek.
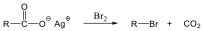
A geminális dihalogénszármazékok előállítására már láttunk az előzőekben példát. Vicinális dihalogénszármazékot olefinekből halogén-addícióval lehet szintetizálni.
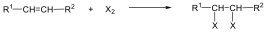
A halogénezett szénhidrogének fizikai tulajdonságai
A halogénezett szénhidrogének forráspontja magasabb, sűrűségük nagyobb a megfelelő szénhidrogénhez viszonyítva. A fizikai tulajdonságok a halogénatomok atomtömegétől és az halogénatomok számától függően változnak. Több halogén beépülésével az olvadás- és forráspont, valamint a sűrűség is nagyobb lesz.
Vízben nem oldódnak, a vizet és a legtöbb szervetlen vegyületet nem oldják, viszont számos szerves vegyületet jól oldanak és a szerves oldószerekkel elegyednek.
A halogénezett szénhidrogének kémiai tulajdonságai
A szén-halogén kötés polarizált és polarizálható. Emiatt a halogénezett szénhidrogének reakcióképesek és sokféle átalakulásra képesek, amelyek nagy része ionos, nukleofil mechanizmussal játszódik le. A halogénatomok nagy elektronegativitása alapállapotban is elektroneltolódást okoz a szén-halogén kötésen és a szénen parciális pozitív, míg a halogénen parciális negatív töltés lesz.
A kötés polározottsága okozza a halogénszármazékok dipólusmomentumát. Kémiai reakció során ez a polározottságot a nukleofil ágens és a poláris oldószer molekulák tovább növelhetik.
Nukleofil szubsztitúciós reakciók
A halogénszármazékok nukleofil szubsztitúciója is kétféle módon játszódhat le. Bimolekuláris (SN2), vagy monomolekuláris (SN1) mechanizmus szerint. Alkil- és vinil-halogenidek esetében, ha primer és szekunder szénatomhoz kapcsolódik a halogénatom, a tipikus reakció SN2, míg tercier szénatomhoz történő kapcsolódás esetén a tipikus reakció SN1.
Az SN2 reakció kinetikailag másodrendű, a reakció sebessége a két reaktáns koncentrációjának szorzatával arányos. A reakcióban az alapállapotban is polarizált szén-halogén kötést a reakcióban résztvevő nukleofil részecske ellenkező irányból közelíti meg és hozzákötődik az elektronszegény szénatomhoz, amelyhez így átmenetileg pentakovalens állapotba kerül. Ezt követően a bekötődő nukleofil részecske kötést alakít ki a szénnel, ezzel egyidőben a halogén anionként lehasad.
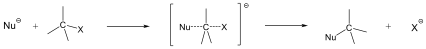
Ha a halogént hordozó szénatom királis volt, a reakcióban a konfigurációja megváltozik. Ezt a jelenséget Walden figyelte meg először, így róla elnevezve Walden-inverzóként ismeretes.
SN1 mechanizmusú elsőrendű reakciók monomolekulárisak, két lépésben játszódnak le. A kétlépéses folyamatok esetén a lassúbb lépés határozza meg a reakció sebességét. Ebben az esetben első lépésben a C-halogén kötés heterolitikus felbomlása történik meg, a második lépésben pedig a nukleofil részecske reagál a karbónium ionnal, ami az első lépéshez képest sokkal gyorsabban zajlik le. Ennek következtében az SN1 reakciók sebességét az első lépés sebessége határozza meg.
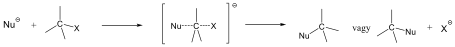
Ha a reakcióban résztvevő halogénatomot hordozó szénatom királis volt, a termékben 1:1 arányban lesz jelen a két enantiomer, tehát racemizáció következik be.
Jelentős szerepet játszik a reakcióhoz alkalmazott oldószer tulajdonsága. Poláris, protikus oldószer elősegíti a C-halogén kötés felhasadását.
SN1 reakció esetén a reagáló nukleofil ágens reaktivitása és koncentrációja nem befolyásolja a reakciósebességet. A kétféle mechanizmust a valóságban nem lehet élesen elválasztani egymástól, többnyire a halogénvegyület szerkezete és a reakciókörülmények szabják meg az egymás mellett lejátszódó folyamatok irányát. Az oldószer fontos szerepet játszik viszont, mert segítheti a disszociációt és a kialakuló kation szolvatációját.
Az oldószereket három csoportba oszthatjuk: protikus, apoláris, dipoláris-aprotikus.
A protikus oldószerek főleg O-H, vagy N-H csoportokat tartalmaznak, nagy a dielektromos állandójuk, hidrogénkötést tudnak kialakítani. Az O-H és N-H csoportok hidrogénjei lehasadhatnak, az oxigén és nitrogén atomok nemkötő elektronpárjaival koordinatív kötéseket is tudnak létesíteni. Ilyen oldószerek pl.: víz, metanol, etanol, hangyasav, ecetsav, formamid.
Az apoláris oldószerek dielektromos állandója kicsi,, nincsenek bennük olyan csoportok, amelyek hidrogénkötést tudnának kialakítani, de koordinatív kötést létesítésére is kevésbé van lehetőség. Ebbe a csoportba tartoznak pl.: benzol, kloroform, dioxán.
Dipoláris-aprotikus oldószerekben nincs olyan csoport, amely hidrogénkötést tudna kialakítani, a hidrogénatomjaik sem szakíthatók le könnyen protonként. Tartalmaznak viszont erősen poláris kötéseket, NO, CN, CO, SO, így dielektromos állandójuk nagy. Ilyen oldószerek pl.: nitro-metán, nitro-benzol, aceton, dimetil-szulfoxid, dimetil-formamid.
Reakciókészség szempontjából szerkezetük alapján a halogénszármazékok az alábbi csoportokba sorolhatók:
-normális reakciókészségűek: a tercier halogénszármazékok kivételével az alkil- és cikloalkil-halogénszármazékok. Jellemző szubsztitúciós reakció: SN2
- csökkent reakciókészségűek: vinil- és aril-halogenidek. Jellemző szubsztitúciós reakció: SN2
- fokozott reakciókészségűek: tercier-alkil- és allil-származékok. Jellemző szubsztitúciós reakció: SN1
Halogén/halogén csere: Az előállításoknál már bemutatott halogén/halogén csere is nukleofil reakció.

A halogénezett szénhidrogének hidrolízise általában alkoholokhoz vezet. Fokozott reakciókészségű származékoknál már víz hatására is bekövetkezik a hidrolízis, normál reakciókészségűeknél lúgoldattal történő melegítés szükséges, míg a csökkent reakciókészségű vegyületeknél a lúg mellett olyan magasabb hőmérsékletet kell alkalmazni, amit nyomás alatt lehet kivitelezni. Bonyolítja a helyzetet, hogy lúgos közegben számítani lehet eliminációra is.

Vicinális dihalogének hidrolízise vicinális-diolok szolgáltat.

Geminális dihalogének hidrolízise geminális-diolokhoz vezet, amelyek oxovegyületté alakulnak át.
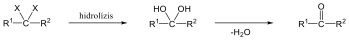
Halogénszármazékok és alkáli-alkohálátok reakciója éterekhez vezet. Pl. metil-jodid és Na-butanolátból butil-metil-éter keletkezik.
A halogénvegyületeket alkoholos oldatban nátrium-hidrogén-szulfiddal melegítve tioalkoholokat adnak.

Nátrium-hidrogén-szulfid helyett nátrium-szulfidot használva tioéterekhez jutunk.

A halogénszármazékok ammóniával is reakcióba lépnek és nem egységes terméket, hanem termékelegyet adnak, amiben különböző rendű aminok találhatók.
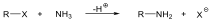
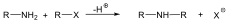
Ha alkáli-cianiddal vizes alkoholos oldatban melegítjük az alkil-halogenideket, akkor karbonsavnitrilekké alakulnak át.

Alkil-halogenidek és karbonsavak alkáli-sóinak reakciójában karbonsavészterek képződnek.

Szénhidrogének előállítási módszere az ún. Wurtz-reakció, amikor alkil-halogenidet alkálifémmel reagáltatnak.

Külön fejezetben tárgyaljuk, de itt is megemlítjük az alkil-halogenidek abszolút éterben fémmagnéziummal való reakcióját, amely egy nagyon fontos, szintetikus szempontból jól alkalmazható ún. Grignard-reagenshez vezet.
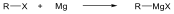
Alkil-halogenideket hidrogénezve gyökös folyamatban dehalogéneződnek. Ezt a reakciót reduktív dehalogénezésnek is nevezik.
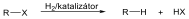
Vinil-haloidok polimerizációja nagyon fontos reakció a polimerek előállításában. A reakció gyökös mechanizmussal játszódik le. Vinil-kloridból kiindulva jutunk a poli-vinil-kloridhoz (PVC).
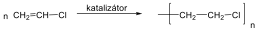
Erős nukleofil ágensek esetében (pl. alkáli-hidroxidok, alkáli-alkoholátok) az előbbiekben részletezett nukleofil szubsztitúció mellett elimináció is bekövetkezhet, a reakcióban az alkil-halogenidből, vagy cikloalkil-halogenidből olefin keletkezik.
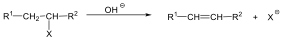
A reakció lehet monomolekuláris (E1) és bimolekuláris (E2).
Az E1 (karbéniumionos) mechanizmus esetén a halogénatom az első lassú, sebességmeghatározó lépésben anionként lehasad, majd a következő lépésben a kation a szomszédos szénatomról protont leadva stabilizálódik. A bruttó reakció monomolekuláris.
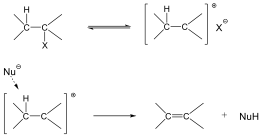
Az E2 mechanizmusú átalakulások során a támadó nukleofil ágens protont hasít le a halogén melletti szénatomról, ezzel egyidejűleg a halogénatom anionként hasad le, tehát közbenső termék keletkezése nélkül megy végbe.
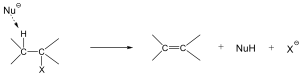
Az E1 és E2 mechanizmus mellett megkülönböztethetünk egy harmadik félét is, az ún. E1cB- vagy karbanionos mechanizmust. Az ilyen folyamat első lépésében reverzibilisen egy karbéniátanion-típusú közbenső termék keletkezik, ami a halogénvegyület konjugált bázisa. A folyamat sebességmeghatározó lépésében ez a konjugált bázis fog elbomlani olefinre és halogenid ionra.
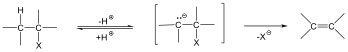
Az E1cB jelölés arra utal, hogy ennek a mechanizmusnak a jellemző lépése a kiindulási vegyület konjugált bázisának monomolekuláris bomlása (c= conjugated, B= base).
A halogénvegyületek esetén az elimináció és a szubsztitúció egymással versengő, ún, kompetitív reakciók és attól függően lesz domináns egyik, vagy másik, hogy milyen a kiindulási vegyület szerkezete és milyenek az alkalmazott reakciókörülmények. Minél magasabb rendű a halogénatomhoz kapcsolódó szénatom, annál inkább az elimináció kerül előtérbe. Hasonló hatása van a nukleofil ágens bázisosságának. Az oldószer polaritásának növekedése viszont a szubsztitúció lehetőségét növeli, mert kedvez a halogéntartalmú molekula disszociációjának, a képződő kation a nukleofil ágenssel tud reakcióba lépni.
Az eddig tárgyalt eliminációs reakciókban a hidrogénatom mindig a β-szénatomról hasadt le proton alakjában, ezért nevezik ezeket a reakciókat β–eliminációnak.
A kloroform és alkálilúgok reakciójában erre azonban szerkezeti okokból nincs lehetőség. Az erős bázis hatására a proton ugyanarról a szénatomról hasad le, mint a kloridanion. Ezt a folyamatot α-eliminációnak nevezzük.

A reakció első lépése gyors, a második lépésben a kloridion lehasadása lassú. A keletkezett igen reakcióképes diklór-karbén lúgos közegben szén-monoxiddá és formiátanionná alakul.
Az aromás gyűrűben, illetve az oldalláncban halogénatomot tartalmazó vegyületek kémiai tulajdonságai különböznek. Ha a halogénatom közvetlenül az aromás gyűrűhöz kapcsolódik, akkor ez az előzőekben részletesen bemutatott alkil-halogenidekhez képest sokkal nehezebben vesz részt nukleofil szubsztitúciós reakciókban. A csökkent reakciókészség az aromás gyűrű –elektronfelhőjének és a halogénatom nemkötő elektronpárjainak kölcsönhatásából adódik.
A klór-benzolt híg lúggal 300 °C-on lehet fenollá hidrolizálni,
míg a para-nitro-klórbenzol esetében ez már szobahőmérsékleten is lejátszódik.
Két reakcióban nem figyelhető meg különbség az alkil- és arilhalogenidek rekaiciókészsége között. Az egyik a gyökös mechanizmus szerint lejátszódó Grignard-reagens (ld. elemorganikus vegyületek) képződése, míg a másik a Wurtz-Fittig-szintézis. Utóbbinál a aril-halogenid és alkil-halogenid elegyét reagáltatják fémnátriummal vízmentes éteres oldatban. Melléktermékként a megfelelő Ar-Ar biaril és R-R alkán keletkezik.

Az alkil-halogenidek és a láncban helyettesített aralkil-halogenidek reakcióinál nincs számottevő különbség.
Fontosabb halogénezett szénhidrogének és felhasználásuk
A klórozott alkánokat ipari méretekben alkánok klórozásával állítják elő. A monohalogén-származékokat alkilezőszernek, főleg mosószerek gyártásánál használják. A polihalogén-származékok fontos oldószerek.
A metil-klorid szobahőmérsékleten gáz, rendkívül toxikus. Ipari előállítása metán klórozásával történik, míg a laboratóriumi méretben metanolból és sósavból cink-klorid jelenlétében állítják elő. A vegyiparban metilezőszerként, illetve hűtőgépekben használják.
A metil-jodid szobahőmérsékleten színtelen folyadék, állás közben megbarnul Viszonylag könnyű kezelhetősége és reakciókészsége miatt a legfontosabb laboratóriumi metilezőszerek egyike. Előállítása dimetil-szulfátból és nátrium-jodidból, vagy metanolból történhet vörös foszfor és elemi jód alkalmazásával.
Az etil-klorid színtelen folyadék, gőze narkotikus hatású. Szerves szintéziseknél etilezőszerként, nagy párolgáshője miatt az orvosi gyakorlatban helyi érzéstelenítőnek használják („fagyasztó”), mert a bőr felületére juttatva gyorsan elpárolog és lehűti, ezzel csökkenti a fájdalomérzetet. Iparilag az etán klórozásával vagy etilénre történő hidrogén-klorid addícióval állítják elő. Laboratóriumban etanolból sósavval cink-klorid jelenlétében lehetséges előállítani.
A ma már hazánkban sem forgalmazott ún. ólmozott benzin oktánszámnövelő adalékként ólom-tetraetilt tartalmazott, amelynek előállítását etil-kloridból valósították meg.
Az etil-bromidot és etil-jodidot etilezőszerként használják. Etanolból állíthatók elő hidrogén-bromid, ill. vörösfoszfor és jód segítségével.
A vinil-klorid szobahőmérsékleten gáz halmazállapotú, erősen toxikus vegyület. A műanyagipar fontos alapanyaga, belőle készül gyökös mechanizmusú polimerizációval a PVC. Kezdetben acetilénből állították elő, ma már etilénbázison történik a gyártása. Az etilénből etilén-dikloridot állítanak elő, majd ezt pirolizálják.
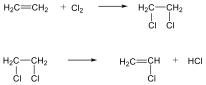
Az allil-kloridot a propén termikus klórozásával állítják elő (ld. „A halogénezett vegyületek előállítása” c. fejezet).
A metilén-diklorid színtelen folyadék, vízzel nem elegyedik, sűrűsége nagyobb a vízénél. Főként oldószerként használják. Vízzel nem elegyedik, sűrűsége nagyobb a vízénél. Iparilag metán klórozásával történik az előállítása.
Az etilén-dikloridot (ClCH2CH2Cl) iparilag etilén klóraddíciójával állítják elő. Felhasználják oldó- és tisztítószerként, továbbá alapanyagként. Legfontosabb felhasználása a magas hőfokon (600 °C) végzett pirolízise vinil-kloriddá.
Az 1,1,2,2-tetraklór-etán, a triklór-etilén és a tetraklór-etilén fontos oldó- és tisztítószerek.
A kloroform színtelen folyadék, vízzel nem elegyedik, nem éghető, sűrűsége nagyobb a vízénél. Előállítása a metán klórozásával történik. Elsősorban oldószerként használják. Jól oldja a zsírokat, így tisztítószerként is alkalmazzák. Régebben sebészi altatóként is használták, de ma már ilyen célra már korszerűbb, kevésbé toxikus anyagokat fejlesztettek ki. Állás közben, elsősorban a levegő oxigénjével foszgénné alakul át, ami illékony és rendkívül toxikus.
A tetraklór-metán (szén-tetraklorid) színtelen, vízzel nem elegyedő, a víznél nagyobb sűrűségű folyadék, amely nem éghető, így kisebb laboratóriumi tüzek oltására is alkalmas. Nagyon jó oldó-, extraháló- és tisztítószer, illetve fluor-klór-metánok előállítására használják. Előállítása metán vagy szén-diszulfid klórozásával történik.
A fluor-klór-metánokat tetraklór-metánból parciális halogéncserével állítják elő. A tetraklór-metánt SbCl5 katalizátorral, vízmentes hidrogén-fluoriddal reagáltatják. A reakcióban difluor-diklór-metán és fluor-triklór-metán lesznek a főtermékek.
A termékelegyből a sósavat vízzel kimossák, az egyes komponenseket frakcionált desztillációval választják el.
A difluor-diklór-metán nem korrozív, nem gyúlékony és nem toxikus. Hosszú ideig hűtőgépekben, légkondicionáló berendezésekben hűtőgázként használták Freon 12 néven. Használatát - az ózonpajzsot károsító tulajdonsága miatt - korlátozták.
A tetrafluor-etilént (perfluor-etilén) ipari méretben a kloroform részleges halogén-cserereakciójával képződő difluor-klór-metán pirolízisével állítják elő.
A perfluor-etilén gyökös polimerizációjával állítható elő a teflon. Ez az anyag kémiailag és termikusan nagyon ellenálló, a háztartásban különböző főzőedények, az iparban reaktorok belső felületének bevonására használják.
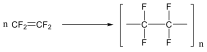
Az illékony halogénezett vegyületeknek narkotikus hatása van, ezért sebészi altatóként használják a korábban említett etil-kloridot, de alkalmazzák az izopropil-kloridot és triklór-etilént is. A fluortartalmú származékok közül a 2-bróm-1,1,1-trifluor-2-klór-etánt (Fluothane, vagy Halothane), és a Enflurane-t használják.
A klór-benzol jellemző, szúrós szagú folyadék. Oldószerként és alapanyagként is felhasználják pl. fenol, anilin, illetve ezekből kiindulva gyógyszerek, színezékek, műanyagok és műszálak előállítására. Előállítása benzol klórozásával történik.
A p-diklór-benzolt molyirtóként használják.
A hexaklór-ciklohexánt (HCH) rovarirtószer, amit benzol klóraddíciójval állítanak elő. A reakcióban különböző izomerek keletkeznek. Rovarirtószerként a γ-izomer a leghatásosabb.
Az 1,1-bisz-(p-klór-fenil)-2,2,2-triklór-etánt (DDT: diklór-difenil-triklór-etán) és több analóg származékot az 1940-es évektől rovarirtóként alkalmaztak, de használatukat hazánkban is több évtizede betiltották, mert kiderült, hogy pl. halakra és madarakra is toxikusak, bekerülve a táplálékláncba az emberekre is veszélyesek. Hosszú lebomlási ideje miatt a DDT felhalmozódott a környezetben és az élőlényekben is.
 |
 |
 |
|
HCH |
γ-hexaklór-ciklohexán |
DDT |
Az eddig ismert vegyületek közül az egyik legtoxikusabb a dioxinnak is nevezett 2,3,6,7-tetraklór-dibenz-dioxin, amely teratogén, mutagén és karcinogén hatása miatt különösen veszélyes az emberre.
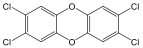
A szerves vegyületekben a szén és hidrogén mellett O, N, S, P és halogének fordulnak elő. Ezek az organogén elemek. Azt a vegyületcsoportot, amelyben az előzőekben említettek mellett más elemeket is tartalmaz kovalens kötéssel a szénatomhoz kapcsolódva, elemorganikus vegyületeknek nevezzük. Ezen belül megkülönböztetjük a fémtartalmú vegyületeket, amelyeket fémorganikus vegyületeknek nevezünk.
Nevezéktan
Elnevezésüknél az általános szabályszerűségek érvényesek. Példaként néhány vegyületet elnevezésükkel együtt bemutatunk.
Elemorganikus vegyületek előállítása
Általános módszerek
A legtöbb szintézis halogéntartalmú szerves vegyületekből indul ki, amit közvetlenül fémmel reagáltatnak.

Erre példa lehet a halogénvegyületek reakcióinál már bemutatott Grignard-reagens előállítása.
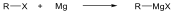
Ha fémorganikus vegyületet reagáltatunk szerves halogénszármazékkal, halogén-fém cserereakció játszódhat le.

Aril-halogenidek ezen az úton átalakíthatók aril-lítiummá, ami közvetlenül fémmel történő reakcióval sokszor nehézségekbe ütközik.

A reakcióban csak a brómatom fog lítiumra cserélődni, mert csak a jód- és brómszármazékoknak van megfelelő reakcióképességük, a klór és fluor esetében nem következik be reakció.
Elemorganikus vegyületek állíthatók elő közvetett módszerrel, amelyben a halogénezett szerves vegyületből Grignard-reagenst készítenek, majd szervetlen halogeniddel reagáltatják. Pl. szilícium-tetrakloridból alkil-klór- és alkil-szilánokat kaphatunk.
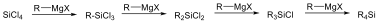
A Grignard vegyületek kémiai tulajdonságai
Az első ilyen származékot 1900-ban Grignard állította elő először, amiért 1912-ben Nobel díjat kapott. A róla elnevezett reagenst vízmentes éterben állítják elő, nem szokás őket izolálni, hanem az éteres oldatot használják fel a további reakciókban.
Szubsztitúciós reakciók során a Grignard-reagens halogéntartalmú szerves vegyületekkel, feszült gyűrűt tartalmazó éterekkel és ortoészterekkel reagálhatnak.
Halogénezett szénhidrogénekből és a megfelelő Grignard-reagensből acetilénhomológokat tudunk előállítani.

Feszült gyűrűt tartalmazó éterek, pl. etilén-oxid, vagy oxetán reakciója Grignard-reagenssel több irányban is hasznosítható. Egyrészt alkoholok állíthatók elő ezen az úton, másrészt az éter szénatom számától függően 2, vagy 3 szénatommal szénláncok meghosszabbíthatók.
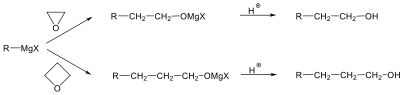
Szerves ortoészterek és Grignard-reagens reakciójában az észter egyik alkoxicsoportja fog kicserélődni a Grignard-reagensben lévő szénhidrogén csoportra.

Grignard-vegyületekkel addíciós reakcióba lépnek a karbonil- és nitrilcsoportot tartalmazó származékok. A képződő vegyületek hidrolízisével oxovegyületből kiindulva alkoholokhoz, nitrilből pedig oxovegyületekhez juthatunk.
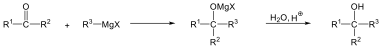
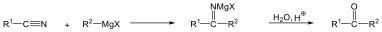
A Grignard-vegyület reakcióba lép szén-dioxiddal is, az előzőekhez hasonlóan képződő addíciós vegyület hidrolízise karbonsavakhoz vezet.
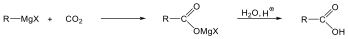
Aktív hidrogént tartalmazó és Grignard-vegyületek között is lejátszódik reakció. Amennyiben az R2 metilcsoport, a képződő metán térfogatos meghatározásán alapulva, alkalmas terminális acetilének kvantitatív mérésére. A módszert Cerevitinov-féle aktív hidrogén meghatározásnak nevezik.

A szilicium tulajdonságai állnak az elemek közül legközelebb a szénéhez. A szilícium is rendelkezik, ha nem is olyan változatossággal mint a szén, hogy láncokat alkosson.
Az előzőekben bemutatott reakció szerint szilícium-tetrakloridból lehet előállítani alkil- és alkil-klór-szilánokat.
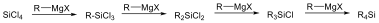
Az alkil-klór-szilánokat elhidrolizálva, a beépült klóratomok számától függően jutunk szilanolokhoz, szilándiolokhoz, vagy szilántriolokhoz, amelyek instabilak és vízkilépés közben kondenzálódnak. Így képződnek a lineáris polisziloxánok, amelyeket kenő- és transzformátorolajként használnak, de szilikonkaucsuk, szilikongyanták is ilyen úton állíthatók elő.
Ólom(II)-halogenidek és Grignard-vegyületek ólomorganikus származékokká alakulnak át.
Ezzel a módszerrel állították elő a már betiltott ún. „ólmozott” benzin adalékát, ami tetraetil-ólom volt (ld. Halogénezett szénhidrogének fejezet).
Ellenőrző kérdések:
-
Hogyan csoportosíthatók a szerves halogénvegyületek?
-
Melyek a szerves halogénvegyületek jellemző fizikai tulajdonságaik?
-
Milyen kiindulási vegyületekből állítható elő etil-klorid? Legalább 2 példát adjon egyenlettel!
-
Írja fel a propán klórral és brómmal való reakcióinak egyenletét!
-
Írja fel az R-X (R=alkil, X=halogén) vegyület reakcióegyenletét ammóniával
-
Írjon fel egy allil-átrendeződési reakciót!
-
Mi a különbség az SN1 és SN2 reakciók között. Adjon rájuk példákat!
-
Mi a különbség az E1 és E2 mechanizmussal lejátszódó eliminációs reakciók között? Adjon meg példákat!
-
Hogyan tud vinil-kloridot előállítani?
-
Adjon példákat csökkent és fokozott reakciókészségű halogénvegyületekre! Mi alapján történik a megkülönböztetés?
-
Milyen atomok és kötések fordulnak elő elemorganikus vegyületekben?
-
Melyek a legismertebb fémorganikus vegyületek?
-
Milyen módszerek ismeretesek fémorganikus vegyületek előállítására?
-
Melyek a Grignard-reagens jellemző reakciói?
-
Milyen reakción alapszik a Cerevitinov-féle aktív hidrogén meghatározás?
-
Írja fel a szerkezeti képletét az alábbi vegyületeknek:
2-brómbután, klórciklopentán, propil-jodid, allil-klorid, 1-fluor-2-metilpropán
-
Mi a helyes kémiai elnevezésük a következő vegyületeknek?
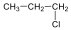 |
 |
 | |
 |
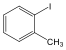 |
Alkoholoknak nevezzük azokat a vegyületeket, amelyekben sp3 hibridállapotú szénhez OH-csoport kapcsolódik.
Enolok azok a származékok, amelyekben a hidroxilcsoport telítetlen, sp2 hibridállapotú szénhez, míg a fenolok esetében a OH-csoport közvetlenül aromás gyűrű szénatomjához kapcsolódik.
Az éterekben két szénatom oxigénatomon keresztül kapcsolódik össze.
Az egyes vegyületcsoportokat, mivel különböznek a kémiai sajátságaik, külön tárgyaljuk.
|
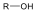
|
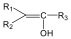
|
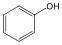
|
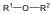
|
|
alkohol |
enol |
fenol |
éter |
Csoportosítás,
Az alkoholokat - a halogéntartalmú vegyületekhez hasonlóan - többféle szempont szerint csoportosíthatjuk:
-
a hidroxilcsoportok száma (egy-, két, három- és több értékű alkoholok),
-
a hidroxilcsoport(ok)hoz kapcsolódó szénatom rendűsége (I. rendű = primer, II. rendű = szekunder, III. rendű = tercier alkoholok),
-
a hidroxilcsoport(ok)hoz kapcsolódó szénváz minősége (telített, telítetlen, nyílt láncú, aliciklusos, aromás gyűrűvel helyettesített),
-
a hidroxilcsoport és a hozzá kapcsolódó szénváz jellemző szerkezeti részlete (telítetlenség, aromás szubsztituens, stb.) közötti távolság,
-
két- és több értékű alkoholok esetében a hidroxilcsoportok viszonylagos helyzete (geminális, vicinális vagy röviden vic- és diszjunkt kétértékű alkoholok stb.).
Példák a különböző alkoholokra:
|
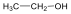
|
|
| |
|
egyértékű |
kétértékű |
háromértékű |
|
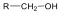
|
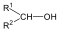
|
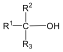
| |
|
I. rendű v. primer |
II. rendű v. szekunder |
III. rendű v. tercier |
|
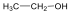
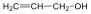
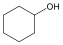
| ||||
|
telített, nyílt láncú |
telítetlen |
telített gyűrűs |
aromás gyűrűs |
|
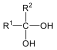
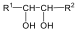
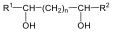
| ||
|
geminális |
vicinális |
diszjunkt (n=1,2,3…) |
Izomériaviszonyok,
A hidroxilcsoport(ok) beépülésének következtében az izomerek száma megnő, az OH-csoportok számának és természetesen a szénlánc hosszának növekedésével ugrásszerűen egyre több szerkezeti izomer lehetséges.
Nómenklatúra
Az alkoholokat a szubsztitúciós nómenklatúra elvei alapján nevezhetjük el. A szubsztitúciós nómenklatúra szerint az alkoholokat a szénhidrogének származékaiként –ol végződéssel nevezzük el. Az elnevezéshez az alkoholok főláncát kell kiválasztani úgy, hogy a leghosszabb normális szénlánc legyen, amely a lehető legtöbb hidroxilcsoportot, ill. telítetlenséget tartalmazza.
|
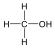
|
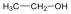
|
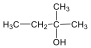
| |
|
metanol |
etanol |
2-metilbután-2-ol | |
|
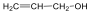
|
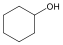
|
| |
|
prop-2-én-1-ol |
ciklohexanol |
ciklohex-2-én-1-ol |
Az oldalláncban lévő több hidroxilcsoportra a megfelelő sokszorozó taggal utalunk.
|
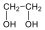
|
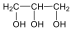
|
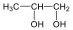
|
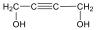
| |
|
etán-1,2-diol (etilén-glikol) |
propán-1,2,3-triol (glicerin) |
propán-1,2-diol |
but-2-in-1,4-diol |
Az alkoholok főláncának számozásakor a hidroxil-csoportok helyzetszámainak, majd a telítetlenségek, ill. az elágazások a számozásának a lehető legkisebbnek kell lennie.
Olyan egyértékű alkoholok esetében, amelyekben a szénhidrogén csoport önállóan elnevezhető, a csoport-funkciós nevek is használhatók, amit a szénhidrogén csoport nevéhez az „alkohol” végződést kapcsoljuk. A metanol ilyen ezen a módon történő elnevezése pl. metil-alkohol, a etanolé etil-alkohol, a ciklohexanolé ciklohexil-alkohol.
A szisztematikus nevek mellett használatosak még a triviális nevek, pl. etilénglikol, glicerin.
Az alkoholok előfordulása és előállítása
A természetben több alkohol fordul elő szabad állapotban is, nagy mennyiségű alkohol viszont kötött állapotban, észterek formájában, amelyek a növényi és állati olajok, zsírok, viaszok főkomponensei.
Egyértékű alkoholok
Az alkoholok ipari, ill. laboratóriumi előállítására sok eljárás ismeretes:
Szintézisgázból, szén-monoxid és hidrogén 1:2 térfogatarányú elegyéből metanol állítható elő.
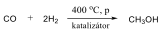
Hosszabb láncú telített szénhidrogének katalitikus oxidációjakor ún. zsíralkoholok keletkeznek.
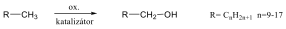
A reakcióban melléktermékként oxidációs termékek, oxovegyületek, zsírsavak is képződnek.
Olefinekből vízaddícióval alkohol képződik. A folyamathoz elektrofil katalizátorra van szükség, leggyakrabban kénsavat használnak. Első lépésben az olefin protonálódásakor karbéniumion képződik, amely a kénsav töménységétől függően alakul tovább alkohollá, vagy kénsav megfelelő savanyú észterévé, amit külön lépésben hidrolizálunk a kívánt alkohollá.
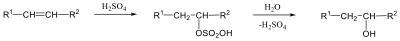
Az olefin szerkezetétől függően különböző rendű alkoholok állíthatók elő. Az addíciónál a Markovnyikov-szabály érvényesül.
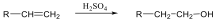
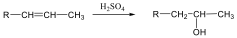
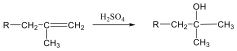
Olefinekből oxidációval kétértékű alkoholok (vic-glikolokat) is szintetizálhatók (ld. kétértékű alkoholok).
Halogénezett szénhidrogénekből hidrolízissel állíthatók elő alkoholok. Ezt a reakciót a halogénszármazékoknál már ismertettük, de meg kell jegyezni, hogy a hidrolízis mellett, főleg III. rendű alkil-halogenidek és vicinális-dihalogén-származékok esetében eliminációs reakció is bekövetkezhet. A keletkezett alkohol rendűségét a halogén atomhoz kapcsolódó szénatom rendűsége szabja meg:
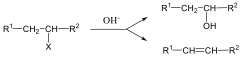
A halogén/hidroxil csere SN2 reakció. A mechanizmusból adódóan, amennyiben a halogén atomhoz kapcsolódó szénatom aszimmetriás, általában megváltozik a konfiguráció, inverziót tapasztalunk (SN1 reakció esetében sokszor racemizáció történik, de különböző tényezők következtében bekövetkezhet inverzó is, sőt maradhat az eredeti konfiguráció is, amit retenciónak nevezünk).
Gyűrűs éterekből, főleg három tagú gyűrűt tartalmazó epoxidokból is lehet kétértékű alkoholokat szintetizálni, de Grignard-reakcióval és Friedel-Crafts reakcióval egyértékű származékok is nyerhetők.

Oxovegyületekből redukcióval viszonylag könnyen állíthatók elő alkoholok. Aldehidekből primer, ketonból szekunder alkohol képződik. Tercier alkoholt ezen az úton nem lehet előállítani. Redukálószernek naszcensz hidrogén, illetve katalitikusan Pt, Pd, Ni jelenlétében hidrogéngáz használható. A komplex fémhidridek közül LiAlH4-t és NaBH4-t használnak, a redukció enyhe körülmények között is lejátszódik.
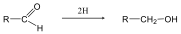

Ha az R-csoport tartalmaz még más, redukcióra érzékeny funkciókat, akkor szelektív redukciós módszereket kell alkalmazni. Ilyen pl. a Meerwein-Ponndorf-Verley-redukció, amelyben az oxovegyületet alumínium-alkoxid (pl.alumínium-izo-propilát) jelenlétében reagáltatjuk, az egyensúlyt az aceton kidesztillálásával eltolható a termékek irányába, majd a képződött alumínium-alkoholáthoz híg vizes savat adva az alkohol felszabadítható.
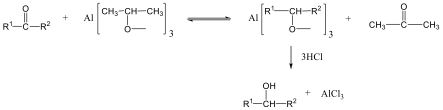
Karbonsavakból és karbonsavszármazékokból (észterekből) történhet. Karbonsavból mindig primer alkoholt lehet így előállítani. Mivel a karboxilcsoport az egyik legnehezebben redukálható funkciós csoport, a reakció katalizátort és erőteljes körülményeket igényel.
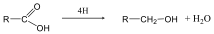
Az észterben lévő alkoholrész a redukció során megtartja az eredeti rendűségét.
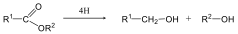
A redukciót hidrogéngázzal pl. réz-kromit katalizátorral lehet végrehajtani, a komplex fémhidridekkel pedig enyhe körülmények között játszódik le a reakció. Utóbbiakkal más karbonsav-származékok pl. savhalogenidek, savamidok, savanhidridek is alkohollá redukálhatók. Észterek fémnátrium/etanol rendszerben is átalakíthatók alkohollá. Lúgos közegben a karbonsav-észterek elhidrolizálhatók, az észteresítő alkohol felszabadítható.
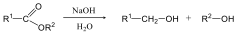
Ipari méretben alkoholokat olefinek és szénmonoxid együttes katalitikus redukciójával is előállítanak, amikor a körülményeket úgy választják meg, hogy a reakció ne álljon meg az aldehidnél. Ezzel a módszerrel I. rendű alkoholokhoz juthatunk.
Oxovegyületekből és észterekből Grignard-vegyületek segítségével is alkoholokhoz juthatunk. Aldehidekből szekunder (formaldehidből primer), ketonokból és észterekből tercier alkoholok állíthatók elő.

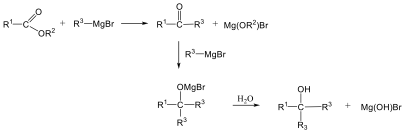
Etilalkohol előállítható szénhidrátokból biológiai úton, erjesztéssel is.
Az alkoholok fizikai tulajdonságai.
Az alkoholok fizikai tulajdonságai átmenetet képeznek az alkánok és a víz között. A rövid szénláncú alkoholok a víz fizikai tulajdonságai tulajdonságaihoz, a hosszabb szénláncúak viszont a szénhidrogénekéhez hasonlítanak. Homológ sorozatokon belül az olvadás-és forrráspontok, a sűrűségek nőnek. Szobahőmérsékleten a legegyszerűbb alkohol a metanol is folyadék. A metánhoz képest a forráspontja 226 °C-kal magasabb, molekulatömegéhez képest kiugróan magas ugyanúgy, mint ahogyan a víz is viselkedik. A molekulatömeg növekedtével az analóg n-alkánok-alkoholok forráspontjának különbsége fokozatosan csökken, egyre inkább hasonlóak lesznek az alkánokhoz
A normális szénláncú egyértékű alkoholok homológ sorozatában a forráspont a szénatomszámmal szabályosan nő, C11-ig szobahőmérsékleten folyadékok, az 1-dodekanol viszont már kristályos. Sűrűségük – függetlenül a szénlánc szerkezetétől – szűk tartományon belül: 0,78 és 0,83 g/cm3 között mozog.
A telítetlen egyértékű alkoholok fizikai tulajdonságai hasonlóak a telített egyértékű alkoholokéhoz, több hidroxilcsoport beépülésével az olvadáspont, forráspont és a sűrűség jelentősen megemelkedik. Az aromás alkoholok is magas olvadás/forrásponttal és sűrűséggel rendelkeznek. Azonos szénatomszámú alkoholok közül az el nem ágazó láncú, primer származékoknak a legmagasabb a forráspontja.
A kis szénatomszámú alkoholok, a metanol, etanol, a két izomer propanol és a III. r. butanol vízzel korlátlanul elegyedik, de a négy szénatomos normális láncot tartalmazó 1-butanol már csak korlátozottan oldódik szobahőmérsékleten vízben. A szénatomszám növelésével az oldhatóság vízben rohamosan csökken, a szilárd halmazállapotúak gyakorlatilag nem oldódnak. A hidroxilcsoportok számának növelése viszont a vízben való oldhatóságot növeli. Szerves oldószerekben általában jól oldódnak. A folyékony halmazállapotú vegyületek jellemző szagúak.
Az alkoholok és a megfelelő szénhidrogének fizikai tulajdonságai közötti különbségeket az alkoholok hidroxilcsoportja(i) okozzák, mivel hidrogénkötések kialakítására képesek.
Ennek eredményeképpen több molekulából álló asszociátumok jönnek létre. Vizes oldatban hidrogénkötések nem csak azonos molekulák között, hanem különböző, pl. alkohol- és vízmolekulák között is létrejöhetnek.
Az egyértékű alkoholok kémiai tulajdonságai
Az alkoholok semleges kémhatású vegyületek, de disszociálnak, a pK értékük 16 körül alakul. Az OH-csoport hidrogénjai ún. aktív hidrogének. A Brönsted-besorolás szerint amfoternek minősülnek, az oxigén nemkötő elektronpárjai Lewis-savakat is képesek megkötni, tehát az alkoholok Lewis-bázisok.
Az aktív hidrogén reakciói:
Fémekkel az alkohol savként reagál. Hidrogénfejlődés közben alkáli-alkoxidok képződnek. A reakció sebességét az alkohol rendűsége befolyásolja, a rendűség növekedésével, a reakció lassul. A fémek minősége is befolyásolja a reakció hevességét. A kálium és nátrium hevesen reagál, míg az alumíniumot aktiválni kell előzetesen.

Alkohol és Grignard-reagens reakciójában szénhidrogén keletkezik. Metil-magnézium-jodid esetében metán fejlődik, amely térfogatosan mérhető. Mivel a módszer kvantitatív, így alkalmas egy vegyületben a hidroxilcsoportok számának meghatározására.

Lítium-alumínium-hidrid és alkohol reakciójában hidrogén fejlődik. Ez a reakció is alkalmas az aktív hidrogéntartalom mérésére.

Az alkoholos hidroxilcsoportokon a disszociáció követeztében protoncsere-folyamat történik, így a hidrogén nehézvízben deutériumra cserélődik.
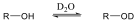
Az észterképzés az alkoholok egyik legfontosabb reakciója, melynek során az alkohol karbonsavval, vagy szervetlen savval lép reakcióba.

Az egyensúlyi reakció katalizátorral, pl. ásványi savval, vagy a képződő víz eltávolításával eltolható az észterképződés irányába.
Karbonsavkloridokkal és karbonsavanhidridekkel az alkoholok ugyancsak észterekké alakíthatók.
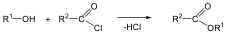
A szervetlen oxosavak közül a salétromsav, ortofoszforsav és kénsav észterei a legfontosabbak, biológiai szempontból kiemelkednek közülük a foszforsav észterek.
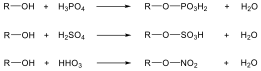
A primer alkoholokat 10 %-os krómkénsavval aldehidekké, kálium-permanganáttal egészen karbonsavig lehet oxidálni.
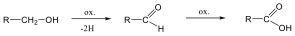
A szekunder alkoholok hasonló körülmények között ketonná alakulnak, majd lánchasadás következik be és karbonsav keletkezik.
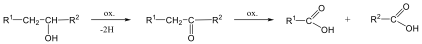
A tercier alkoholokat többnyire csak erélyesebb körülmények között lehet oxidálni, amikor lánchasadást követően alacsonyabb szénatom számú keton és karbonsav elegye keletkezik. A képződő keton újabb lánchasadással továbbalakul karbonsavakká.
Iparilag a különböző rendű alkoholok oxovegyületekké, vagy karbonsavakká oxidálhatók gázfázisban a levegő oxigénjéval palládium, réz, vagy réz-oxid katalizátorral
Az alkoholok oxidációja az élő szervezetben is lejátszódik. Az etanol, főleg a májban található dehidrogenázok hatására acetaldehiden keresztül acetáttá alakul. Metanol esetében ugyanezek az enzimek a mérgező formaledehiden keresztül formiáttá oxidálják a metanolt, ami csökkentve a vér pH értékét, acidózis kialakulásához vezet.
Az alkoholok egy feleslegben alkalmazott keton és alumínium-tercier-alkoholát jelenlétében - ún. Oppenauer-oxidációval - szelektíven oxovegyületekké alakíthatók át.

A reakció alkalmazható telítetlen alkohol esetében is, amivel telítetlen oxovegyületek állíthatók elő.
Hidrogén-halogenidek és alkoholok reakciójával alkil-halogenidekhez jutunk. Az alkoholok reakciókészsége rendűségükkel együtt nő. A tercier alkoholok szobahőmérsékleten is átalakulnak, míg primer alkohol és sósav reakciójakor a melegítés mellett még ZnCl2-ra is szükség van
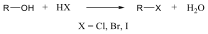
Az alkoholok szervetlen savhalogenidek (pl. PBr3 vagy SOCl2) segítségével is halogénezett szénhidrogénekké alakíthatók.

Alkoholok reagálnak ammóniával. A reakció fontos amin előállítási módszer, pl. metanol alumínium-oxid katalizátoron megfelelő hőmérsékleten és nyomáson különböző rendű metilaminok egyensúlyi elegyéhez vezet.
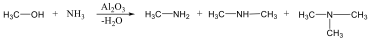
Alkoholokból ún. intramolekuláris vízkilépéssel olefinek, míg intermolekuláris eliminációval éterek keletkeznek. Ha kénsavba 140 °C-nál magasabb hőmérsékleten etanolt csöpögtetünk, akkor etilén keletkezik.
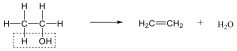
Ha 140 °C alatti hőmérsékleten reagáltatjuk az etanolt kénsavval, ügyelve, hogy az alkohol mindig fölöslegben legyen, akkor dietil-éter képződik.
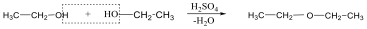
A két reakcióút a reakciókörülményeken kívül az alkohol szerkezetétől is függ. A kisebb szénatomszámú és I. rendű alkoholok inkább éterekké, a nagyobb szénatomszámúak és magasabb rendű alkoholok inkább olefinekké alakulnak át.
Kétértékű alkoholok
Kétértékű alkoholok előállítása
Csak azokat az előállítási módszerek és kémiai tulajdonságok kerülnek részletezésre, amelyek eltérnek az előzőekben ismertetett egyértékű alkoholokétól.
Geminális diolok
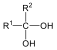
A geminális diolok instabil vegyületek, vízkilépéssel aldehidekké, vagy ketonokká alakulnak át. Stabilabbak azok a vegyületek, amelyekben a hidroxilcsoportok melletti szénatomon elektronszívó tulajdonságú atom(ok), csoport(ok) találhatók.
Vicinális diolok
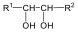
A vicinális diolok általában olefinekből állíthatók elő közvetlenül oxidálószerekkel (pl. KMnO4, H2O2, OsO4). Gyűrűs olefinek esetében ezekkel az oxidálószerekkel cisz-helyzetbe épülnek be a hidroxilcsoportok.
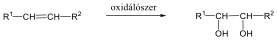
Ólom-tetraacetáttal végrehajtva a reakciót először az 1,2-diol diacetátja keletkezik, ami elhidrolizál 1,2-diollá, amelyben a két hidroxilcsoport transz-térállású lesz.

További lehetőség olefinekből vicinális diolok előállítására, ha az olefinre brómot, vagy hipoklórossavat addícionálunk, majd a képződő dibróm-, vagy klórhidrin származékot elhidrolizáljuk.
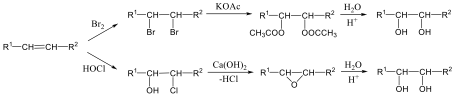
Epoxidokon keresztül úgy is előállítható az 1,2-diol, hogy az olefint persavval epoxidáljuk, amit az előzők szerint elhidrolizálunk. Persavként perhangyasav, perecetsav és perbenzoesav is használható.
Fizikai tulajdonságok
A vicinális diolok sűrű, vízben oldódó folyadékok, amelyeknek édeskés ízük van. Forráspontjuk az azonos szénatomszámú egyértékű alkoholokkal összevetve jóval, akár 100 °C-kal is magasabb.
Kémiai tulajdonságok
A kétértékű alkoholok kémiai tulajdonságai nagyrészt hasonlóak az egyértékű alkoholokéhoz, az észter-, éter- és alkoholátok képződése vonatkozásában, de átalakíthatók gyűrűs éterekké is. Pl. az etán-1,2-diolból 1,4-dioxán állítható elő, ha a diolt híg foszforsavval, vagy kénsavval melegítjük.
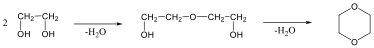
A fenti reakció első lépése ismétlődhet, ami poliéterlánc kialakulásához vezet.
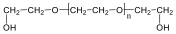
A vicinális diolok oxidációjának terméke(i)t az határozza meg, a hidroxilcsoportok milyen rendű szénatomokhoz kapcsolódnak, de általában termékelegy keletkezik, amiben különböző mértékben oxidált hidroxi-oxo-, oxo- és karbonsavak lesznek.
Vicinális diolok esetében, ha legalább az egyik, a hidroxilcsoportokat hordozó szénatomhoz alkilcsoport kapcsolódik, bekövetkezik az ún. pinakol-pinakolon átrendeződés, amit előszőr ditercier-diolon figyeltek meg. A diolból kénsav hatására lejátszódó vízelvonáskor egy átrendeződés játszódik le a szénvázon és egy oxovegyület keletkezik.
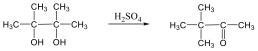
Diszjunkt diolok
n>=1
Diszjunkt diolok előállíthatók halogénszármazékokból lúgos hidrolízissel.
Kémiai tulajdonságaik hasonlítanak a vicinális diolokéhoz azzal a kivétellel, hogy itt nem jellemzőek az oxidatív és átrendeződési reakciók.
Többértékű alkoholok
A háromértékű alkoholok legfontosabb képviselője a glicerin, ami a természetben zsírokban és olajokban különböző karbonsavakkal képzett észterekben fordul elő.
Előállítani ezekből a zsírokból, olajokból is lehetséges, ha elhidrolizáljuk őket nátrium-, vagy kálium-hidroxiddal. A képződő zsírsav alkálisói a szappanok, a másik termék pedig a glicerin.
Szintetikus úton először propilénből allil-kloridot állítanak elő, amit kalcium-hidroxiddal epiklórhidrinné alakítanak tovább, ebből pedig vizes nátrium-hidroxid hatására képződik a glicerin.
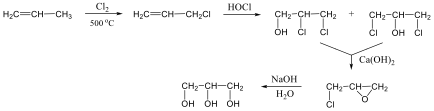
A glicerin kémiai tulajdonságai a vicinális diolokéhoz hasonlítanak.
Mindhárom hidroxilcsoportja karbonsavakkal észteresíthető, a képződő származékok a mono, di- és trigliceridekek.
A glicerint lehet észteresíteni szervetlen oxosavakkal is, így pl. salétromsavval történő reakciója glicerin-trinitrátot szolgáltat, amelyet robbanószerként használnak.
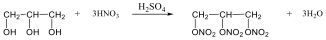
Glicerint hevítve, vagy vízelvonószerekkel dehidratálódik és akrolein képződik.
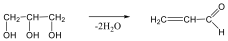
Enolok
Az enolok általában nem stabil vegyületek, a vinilalkoholnak is csak észter- és éter származékai stabilisak. Szabad formájában az ún. enol-oxo-tautoméria következtében, acetaldehiddé alakul át.
Az egyensúly teljesen (100%) az oxo-forma irányába van eltolódva. Ismert a jelenség az oxovegyületek és enolok körében, amikor a szerkezeti adottságok befolyásolják a két forma arányát és bizonyos esetekben stabilizálják az enol-formát. Ez az izomériának egyik fajtája, amikor a két izomer csak egy kettőskötés és egy hidrogén helyében különbözik. Az ilyen típusú, egymásba spontán átalakuló izomereket tautomereknek nevezzük. Megnövekszik az enolforma stabilitása pl. a 1,3-dioxo-vegyületek esetében, amikor molekulán belül, azaz intermolekuláris hidrogénkötés tud kialakulni és a létrejövő kelátgyűrű azt eredményezi, hogy a tiszta 2,4-pentándionban szobahőmérsékelten kb. 80 % az enolforma.

A stabilis enolok közé sorolhatjuk a fenolokat is, de ezt a vegyületcsoportot külön tárgyaljuk.
Az enolokban az OH-csoportban a kötés polaritása megnövekszik az alkoholos hidroxilcsoporthoz képest, mert oxigén nemkötő elektronpárja az enolos szénatom -kötésével konjugációba lép. Ez csökkenti a hidroxilcsoport oxigénjének elektronsűrűségét, végső soron az enolok a víznél erősebb savak. Az enolok savasságát jól példázza az aszkorbinsav.
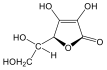
A nagyobb mértékű disszociáció az oka, hogy az enolok híg lúgokban oldódnak, míg az alkoholok csak egy bizonyos szénatomszámig.
Enolok alfa-szubsztitúciója: az enolok a C=C kettőskötéshez hasonlóan reagálnak elektrofilekkel, a reakció tulajdonképpen egy alfa-szubsztitúció.
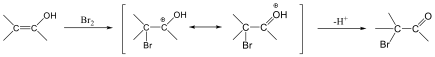
Az enolok fémionokkal élénk színreakciót adnak, pl. Cu2+, Fe3+, Ni2+ ionokkal.
Az alkoholok néhány fontosabb képviselője
Metanol
A növényvilágban is megtalálható, főleg kötött, észterek, éterek formájában. Széles körben használják laboratóriumi és a szerves vegyipari szintéziseknél, az egyik legfontosabb oldószer, de nagy mennyiségben különböző metilezőszereket (dimetil-szulfát, metil-halogenidek), nátrium-metilátot, formaldehidt állítanak elő belőle. Színtelen, vízzel korlátlanul elegyedik. Az etilalkoholhoz hasonló a szaga, veszélyes azonban összetéveszteni, mert nagyon mérgező. Már fél deciliter az emberben vakságot, súlyosabb esetben halált okozhat. Előllítása régen a fa száraz lepárlásával történt, innen ered a faszesz elnevezés. Ma iparilag szintézisgázból, illetve főleg metán parciális oxidációjával történik.
Az etanol ugyancsak fontos oldószer, de élvezeti szerként ősidők óta ismeretes. Nagy mennyiségben használják szintéziseknél, de egyre többet forgalmaznak természetes eredetű etanolt benzinnel keverve motorhatóanyagként (E85). A gyógyászatban fontos fertőtlenítőszer. Ipari előállítása az etilén gőzfázisban végrehajtott hidratálásával történik.
Biológiai erjesztésessel is előállítható pl. gyümölcsökből. Poliszacharid tartalmú alapanyagok is felhasználhatók, de ezeket előbb egyszerű cukorrá kell alakítani. Az átalakulás során széndioxid fejlődik, ami pl. a must forrása alatt felgyűlhet a pincében (mustgáz) és óvatlanság esetén fulladást okozhat.

Az etanol vízzel azeotropot képez, ezért vízmentesítése frakcionált desztillációval nem lehetséges. Abszolút etanolt vízmegkötő szerekkel (pl. Ca, CaO, molekulaszita), vagy benzol hozzáadásával végrehajtott azeotrop desztillációval nyerhetünk.
A propán-1-ol és propán-2-ol oldószerként, szintéziseknél kiindulási anyagként használatos.
Az etilénglikol sűrű, édes ízű folyadék. Iparilag az etilén-oxid savas katalizátorok jelenlétében végzett hidrolízisével állítják elő. Nagy mennyiségekben használják fel autómotorokban fagyálló hűtőfolyadékként.
A glicerin (propán-1,2,3-triol) édes ízű szirupszerűen sűrű folyadék. A növényi és állati zsírokban fordul elő és az előzőekben ismertetett módon belőlük hidrolízissel kinyerhető. A cukrok alkoholos erjedésének közbenső terméke. Nem toxikus, nagy mennyiségben használják az élelmiszer-, gyógyszer- és kozmetikai iparban. A glicerin nitrálásával előállított glicerol-trinitrát robbanóanyagként, helytelen elnevezéssel, mint „nitroglicerin” ismeretes, ami a dinamit alapanyaga, de fontos értágító hatóanyag, oldja a simaizmok görcsét.
A zsíralkoholok az egyértékű C10-C18 szénatomszámú, el nem ágazó láncú alkoholok. Zsírok katalitikus hidrogénezésével állíthatók elő. Kénsavval savanyú észterekké átalakítva szintetikus mosószerként használatosak.
Fenoloknak nevezzük azokat a vegyületeket, amelyekben egy vagy több hidroxilcsoport közvetlenül aromás gyűrű szénatomjához kapcsolódik. Csoportosításuk is a bennük lévő hidroxilcsoportok számán alapszik, így megkülönböztetünk egy-, két-, háromértékű fenolokat.
Nómenklatúra
Az alapvegyület a fenol, vagy hidroxi-benzol. A legegyszerűbb fenoloknak van triviális nevük, ezeket is használhatjuk.
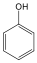 |
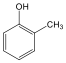 |
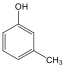 |
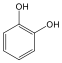 | |
|
fenol |
o-krezol (2-metilfenol) |
m-krezol (3-metilfenol) |
pirokatechin | |
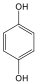 |
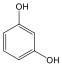 |
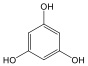 |
 | |
|
hidrokinon |
rezorcin |
pirogallol |
pikrinsav |
A IUPAC nómenklatúra értelmében elnevezésükkor az alapszénhidrogén nevéhez az -ol végződést fűzünk, a megfelelő sokszorozótagot hozzátéve. Így a pirogallol szisztematikus neve 1,3,5-benzoltriol, de a pikrinsavat 2,4,6-trinitro-fenolnak kell elnevezni. A vegyületek számozásánál a legkisebb helyzetszámot a fenolos hidroxilcsoport kapja.
A fenolok előállítása
Fenol és származékai a kőszénkátrány egyes frakcióiban is megtalálhatóak, fizikai és kémiai módszerekkel kinyerhetők. Savas karakterüket kihasználva, lúgokkal kioldhatók, majd a lúgos oldatok megsavanyítása után kiválnak, vagy extrahálhatók.
Fenolokat a megfelelő aromás halogénszármazékokból kiindulva elő lehet állítani, de általában csak erélyes reakciókörülmények alkalmazásával valósítható meg a hidrolízis.

A fenol az ún. Raschig-féle szintézissel állítható elő, melynek első lépésében benzolból oxiklórozással klór-benzolhoz jutnak, amit utána a képződő vízzel egyből elhidrolizálnak. A második lépésben képződő sósavat pedig visszaforgatják az első lépésbe.
Diazóniumsókat vizes oldatban savasan elhidrolizálhatunk, amikor azok melegítés hatására nitrogént veszítenek és fenol képződik.
A diazóniumsókat aromás aminokból diazotálással lehet előállítani (ld. aminok reakciói).
Aromás szulfonsavakat, pontosabban ezek alkálifém sóját, alkalikus közegben megömlesztve a megfelelő fenol alkálifém sójához juthatunk, amiből a fenol savval felszabadítható.
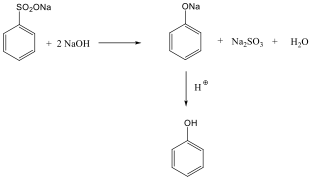
Bizonyos esetekben a szulfonsavak alkáliömlesztése során átrendezett termékek keletkezhetnek.
Iparilag a fenolt benzolból és propilénből nyerhető kumolból kiindulva állítják elő, amihez a kumolt peroxiddá oxidálják, majd elhidrolizálják kénsavval. A fenol mellett aceton keletkezik.
A Dow-eljárás toluolból indul ki, amit első lépésben kobalt-só jelenlétében levegővel benzoesavvá, második lépésben pedig réz és magnézium-só katalizátor alkalmazásával fenollá oxidálnak.
A fenolok fizikai tulajdonságai és élettani hatásuk
A fenolok között vannak szobahőmérsékleten folyékony származékok, de sok fenol kristályos. Molekulatömegükhöz viszonyítva a forráspontjuk magas. Teljesen tiszta állapotban színtelenek, hosszabb tárolás során sötét színűvé válnak. Vízben kis mértékben oldódnak és ők maguk is korlátozott mértékben oldanak vizet. A hidroxilcsoportok számának növekedésével a vízben való oldékonyságuk növekszik. Jellemző szagú anyagok, erősen toxikusak, a kolloid fehérjeoldatokat kicsapják, a bőrt megmarják, sőt a bőrön át is felszívódnak. Több fenolszármazék használatos fungicid (gombaölő), baktericid (baktériumölő), bakteriosztatikus (baktériumszaporodást gátló) és algaellenes szerként.
A fenolok kémiai tulajdonságai
A fenolok reakcióképes vegyületek. Mivel a fenolokban a hidroxicsoport közvetlenül aromás gyűrű szénatomjához kapcsolódik, különbséget tapasztalunk az fenolos- és alkoholos hidroxil csoport kémiai tulajdonságaiban. Másrészt az aromás gyűrű és a hidroxilcsoport befolyásolja egymás reakciókészségét.
A fenolok savassága
A fenolok savas tulajdonságúak, mert OH-csoportjuk könnyen disszociál. Savasságuk az alkoholokénál jóval nagyobb. A fenol pK értéke 10,0, míg pl. a metanolé 15,5, az etanolé 15,9. A különböző fenolszármazékok savi erősségét az aromás gyűrűhöz kapcsolódó szubsztituensek befolyásolják. Pl. 2 db nitrocsoport beépülése esetén a fenolszármazék pK értéke eléri a szerves karbonsavak tartományát, 3 nitrocsoport, mint pl. a 2,4,6-trinitro-fenol (pikrinsav) esetében pedig megközelíti az ásványi savakét.
Az alkoholokéhoz viszonyított erősebb savi tulajdonság az aromás gyűrű jelenlétének következménye, mert a deprotonálódáskor képződő negatív töltés delokalizálódhat. Ha az aromás gyűrűhöz elektronszívó tulajdonságú csoportok kapcsolódnak, a fenolátion stabilitása növekszik. Az előző példákban szereplő nitrocsoport –K effektusa növeli meg jelentős mértékben a fenol savasságát.
A fenolok sóit fenolátoknak nevezzük, a fenol nátriumsója a nátrium-fenolát, amiből a fenol már szénsavval is felszabadítható.
nátrium-fenolát
A fenolok reakcióit két csoportra oszthatjuk és ennek megfelelően tárgyalhatjuk. Elsőbe a fenolos hidroxilcsoport, másodikba az aromás gyűrű reakciói tartoznak.
A fenolos hidroxilcsoport reakciói
A fenolok és anionjaiknak oxigénatomján két, ill. három osztatlan elektronpár van, ezért a fenolok és anionjaik – ugyanúgy, ahogy az alkoholok is – nukleofil reagensek, de a fenolokat ambidens nukleofileknek kell tekintenünk.
A fenolok alkálifémmel alkotott sói (fenol lúg jelenlétében) alkil-halogenidekkel, vagy dimetil-szulfáttal ugyanúgy reagálnak, mint az alkoholok bázis jelenlétében. Első esetben alkil-aril-éterek keletkeznek, míg utóbbi esetében a termék aril-metil-éter.

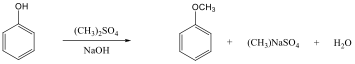
A fenolok speciális alkilezőszerei a diazo-alkánok. A diazo-metánnal történő reakcióban aril-metil-éterek keletkeznek.
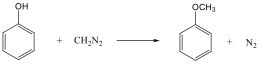
A fenolokat aralkilezni is lehet, mely reakciók közül a benzilezés az egyik legfontosabb, mert a képződő benzil-éterből a fenol katalitikus hidrogénezéssel könnyen visszanyerhető, így a benzilezés felhasználható a fenolos hidroxilcsoportok védőcsoportjaként. A benzilezést általában benzil-kloriddal végzik kálium-karbonát jelenlétében.
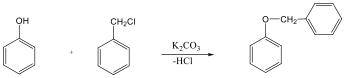
A fenolok közvetlen O-acilezése karbonsavakkal nehezebben játszódik le, mint az alkoholok közvetlen acilezése. Könnyebben acilezhetők a fenolok piridin jelenlétében karbonsav-halogenidekkel.
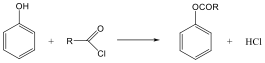
Hidrolízisre nem túl érzékeny (pl. aromás) sav-kloridokkal a fenolok alkálisói vizes-lúgos közegben is acilezhetők (Schotten-Baumann-féle acilezés).
Széles körben használatos még acilezésükre a fenolok forralása savanhidridekkel kis mennyiségű bázikus, vagy savas katalizátor jelenlétében. Fenolból kiindulva ecetsav-fenilésztert kapunk.
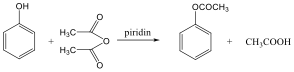
Az ecetsav-fenilészterrel AlCl3, ZnCl2, vagy BF3 jelenlétében intramolekuláris Friedel-Crafts-reakció játszódik le és átrendeződik orto- és para-hidroxi-acetofenonná.
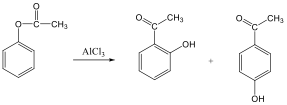
A reakció nemcsak ecetsav-fenilészter esetében játszódik le, hanem általános reakciója a fenolésztereknek, amit Fries-átrendeződésnek nevezünk.
A hidroxilcsoport, deprotonált formájában méginkább aktiváló hatású, orto és para helyzetekbe irányító I. rendű szubsztituens. Így a benzolhoz képest az aromás elektrofil szubsztitúció a fenolokon gyorsabban és enyhébb körülmények között mennek végbe.
A fenolok aromás gyűrűjének halogénezése még Friedel-Crafts-katalizátorokat sem igényel könnyen végbemegy. A fenol klórral való reakciója orto- és para-klór-fenolhoz vezet
A fenol híg vizes oldatban történő brómozáskor pillanatszerűen reagál és 2,4,6-tribróm-fenol keletkezik.
A képződő 2,4,6-tribróm-fenol továbbreagál és egy nem aromás vegyületté, a tribróm-fenol-brómmá alakul, ami egy vízben rosszul oldódó csapadék. Mivel kvantitatívan képződik, analitikai célokra is használható, pl. a fenol kimutatására és meghatározására, illetve analitikai meghatározásoknál alkalmazott brómfölösleg megkötésére.
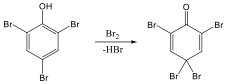
A fenol brómozási terméke függ a reakciókörülményektől is, pl. az oldószer is befolyásolja a reakciót, mert pl. szén-diszulfidban végrehajtva a brómozást, a p-bróm-származék izolálható.
A naftolok monobrómozása 1-naftolból a 4-bróm-1-naftol, ill. 2-naftolból 1-bróm-2-naftol keletkezik.
A fenol nitrálása nem igényli nitrálósav alkalmazását; a mononitro-származékok (orto- és para-nitro-fenol elegye) már szobahőmérsékleten, 40%-os salétromsavval is előállítható. végbemegy és (31) elegyéhez vezet.
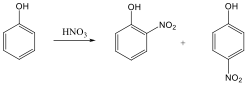
A két izomer nitro-fenol fizikai tulajdonságai számottevően különböznek, mert az orto-izomerben molekulán belüli hidrogénkötés jön létre, ami a para-izomerben, térbeli okok miatt nem alakulhat ki.
A naftolok esetében is könnyen lejátszódik a nitrálás, az észlelt irányítás azonos a mono-brómozás során tapasztalhatókkal.
A fenol kénsavval már szobahőmérsékleten reagál orto-szulfonsav keletkezik, magasabb hőmérsékleten, 100 °C-on pedig a para-izomer képződik. Füstölgő kénsav és magasabb hőmérséklet hatására di-, illetve triszulfonsav keletkezik.
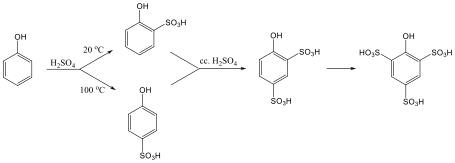
A fenol lúgos körülmények között reagál a formaldehiddel és hidroxi-metilcsoport(ok) épül(nek) be a molekulába.
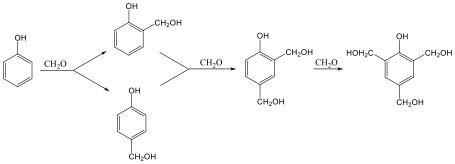
Ezek a vegyületek polikondenzációra hajlamosak. Fenol-formaldehid elegyet melegítve egy gyantaszerű anyag keletkezik, amit tovább hevítve - miközben a molekulákat metilénhidak kötik össze – láncszerű polimer jön létre, majd keresztkötések is kialakulnak. Így hőre nem lágyuló, térhálós szerkezetű anyag keletkezik, amely bakelit néven ismert és széles körben alkalmazzák. A bakelithoz hasonló módon, különböző fenolokból előállított műanyagokat fenoplasztoknak nevezzük.
Hasonló reakciót figyelhetünk meg fenol és aceton között sósav jelenlétében. A keletkező un. biszfenol-A-t polikarbonátok előállítására használják.
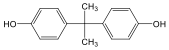
biszfenol-A
A fenolok aromás gyűrűjében nagy elektronsűrűség jön létre a gyűrű és a hidroxilcsoport(ok) konjugációja miatt, ami oxidációra is érzékennyé teszi a fenolok aromás gyűrűjét.
A könnyen végbemenő oxidálódás szemmel is nyomon követhető, mert állás közben az eredetileg színtelen vegyületek megszínesednek. Különösen könnyen oxidálódnak a több hidroxilcsoportot tartalmazó fenolok.
A hidrokinon és a pirokatechin típusú difenolok enyhe oxidációjában kinonok keletkeznek.
A reakció visszafelé is könnyen lejátszódik: a kinonok két hidrogén felvéve a megfelelő fenollá alakulnak. A reakcióval aromás rendszer jön létre, emiatt megy végbe könnyen a hidrogénfelvétel.
A fenolokat vízmentes cink-klorid jelenlétében ammóniával melegítve, hidroxilcsoportjuk aminocsoportra cserélhető. A reakciót a több értékű fenolok esetében alkalmazzák, mert ezeknél részleges aminolízissel amino-fenolok állíthatók elő.
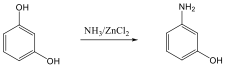
Jellemző reakciója a fenoloknak, hogy vas(III)-kloriddal élénk színreakciót ad oxidáció és/vagy komlexképződés következtében. A szín függ a a hidroxilcsoportok számától és viszonylagos helyzetüktől is.
A fenol katalitikus redukcióval ciklohexanolon keresztül ciklohexanonná alakítható át.
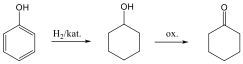
Fontosabb fenolok
A fenol a vegyületcsalád legegyszerűbb és egyben legfontosabb képviselője, tiszta állapotban színtelen, jellegzetes szagú kristályos anyag. Állás közbeni oxidatív folyamatok miatt a színe mélyül. Vízben kis mértékben, alkoholban és éterben jól oldódik. A fenol és általában a fenolok erős sejtmérgek, bőrön keresztül is felszívónak, a velük való munka fokozott figyelmet és óvatosságot igényel. A fenol vizes oldata volt az első antiszeptikum; amit 1867 óta használtak karbolsav néven az orvosi gyakorlatban.
Baktériumölő szerként, fungicidként (gombaölő), algaellenes szerként használják a fenolokat, pl. faszerkezetek, vasúti talpfák, villanyoszlopok impregnálására.
A fenol előállítása kőszénkátrányból, az ismertetett Raschig- és Dow-eljárással, kumolból kiindulva, ill. benzol-szulfonsavból alkáliömlesztéses módszerrel történik. Az előző alkalmazásokon túlmenően nagy mennyiségben használja fel a gyógyszer-, festék- és műanyagipar. Nagy mennyiségben alakítják szalicilsavvá, amit a gyógyszeripari felhasználás mellett az élelmiszeriparban is alkalmaznak. Jelentős volumenben először ciklohexanonná, majd adipinsavvá és kaprolaktámmá alakítják, amelyek a műszálak előállításához használnak.
|
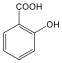
|
|
|
|
szalicilsav |
adipinsav |
kaprolaktám |
A pentaklór-fenol gombaölő és algaellenes szer. A fenol klórozásával állítható elő.
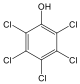
A pikrinsav sárga kristályos anyag, amit a preparatív szerves kémiában különböző vegyületek (főleg aminok és kondenzált gyűrűs aromás szénhidrogének) kristályos származékaik alakjában történő leválasztására és azonosítására használják.
A polifenolok jelentős antioxidáns hatással rendelkező vegyületek, amelyek zöldségekben és gyümölcsökben fordulnak elő. Fogyasztásuk elősegíti az emberi szervezetben az oxidatív stressz elkerülését. Igazoltan az oxidatív stressz következménye számos betegség kialakulása. Az oxidatív stressz akkor alakul ki, amikor a szervezetben a gyökös folyamatokat egyensúlyban tartó szabályozó mechanizmus nem működik megfelelően és megnő a káros szabadgyökök száma.
A vörösborokban jelenlévő rezveratrol ugyancsak ebbe a vegyületcsaládba, a polifenolok közé tartozik.
rezveratrol
Az éterek C-O-C kötést tartalmaznak. Levezethetők vízből a két hidrogénatom, illetve alkoholok és fenolok hidroxilcsoportjában lévő hidrogénatom alifás, vagy aromás oldallánccal történő helyettesítésével, de lehet az éteres oxigén egy gyűrűrendszer tagja is.
 |
 |
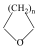 | |
|
éterkötés |
nyílt láncú éter |
gyűrűs éter |
Csoportosítás
Az étereket, az éteres oxigénhez kapcsolódó csoportok szerint is osztályozhatjuk, amelyek lehetnek alkil, cikloalkil-, alkenil, vagy aromás csoportok. Lehetnek az éterek nyíltláncúak és gyűrűsek. A nyíltláncúak esetében megkülönböztetünk egyszerű és vegyes étereket. Amennyiben az oxigénhez kapcsolódó csoportok azonosak, akkor egyszerű éterekről, amennyiben a két csoport különböző, vegyes éterekről beszélünk. Enoléterek azok a származékok, amikor az oxigén kettős kötésben résztvevő szénatomhoz kapcsolódik.
Nevezéktan
Az éterek csoportfunkciós nómenklatúra szerint elnevezése az alábbi példákon keresztül könnyen áttekinthető. Az oxigénatom által összekötött két csoport nevét abc-sorrendben felsoroljuk, majd hozzákötjük az -éter végződést.
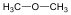 |
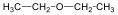 |
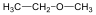 |
|
dimetil-éter |
dietil-éter |
etil-metil-éter |
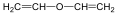 |
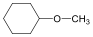 |
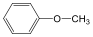 |
|
diviniléter |
ciklohexil-metil-éter |
fenil-metil-éter |
Ezt az elnevezést akkor alkalmazzuk, amikor a csoportoknak önálló neve van. A többi származékot a szubsztitúciós nómenklatúrát alkalmazva nevezzük el a megfelelő szénhidrogének alkoxi, ariloxi-, stb. származékainak.
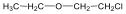 |
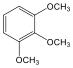 | |
|
1-etoxi-2-klór-etán |
1,2,3-trimetoxi-benzol |
A gyűrűs éterek egyik fontos csoportja az epoxidoké, amelyek az etilénoxid analógjainak tekinthetők.
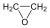 |
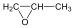 | |
|
etilén-oxid v. oxirán |
propilén-oxid |
A gyűrűs étereket elnevezhetjük epoxi-, vagy heterociklusos vegyületként is az alábbi példa szerint.
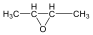
2,3-epoxi-bután v. 2,3-dimetil-oxirán
Előállítás
Éterek a megfelelő alkoholokból intermolekuláris vízelvonással előállíthatók. A reakciót az alkoholok reakcióinál részleteztük.
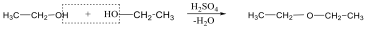
Vízelvonószer lehet tömény kénsav, foszforsav és bórsav is. A reakció magasabb hőmérsékleten kontakt katalizátorral is végrehajtható (pl. Al2O3).
Kétértékű alkoholokból gyűrűs éter is előállítható. Bután-1,4-diolból foszforsavval 70 bar nyomáson és 260 °C hőmérsékleten tetrahidrofurán állítható elő.
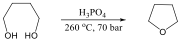
Vegyes éterek a Williamson-féle szintézissel állíthatók elő. A módszer szerint alkil- halogenidek, vagy dialkil-szulfátok alkoxidionnal reagálva, SN2 mechanizmus szerint étert eredményeznek. A módszer alkalmas telítetlen éterek előállítására is.

Fenolok alkálisói alkil-halogenidekkel aromás éterekké alakíthatók át.

A fenolok kémiai tulajdonságainál bemutatott módon a fenolok diazometánnal reagálnak és aromás metil-étereket adnak, hasonlóan mint a fenolok alkálisói dimetil-szulfáttal melegítés hatására.
Alkil-vinil-éterek acetilénre történő alkohol addícióval állíthatók elő-

A gyűrűs éterek háromtagú származékai, az epoxidok olefinekből szintetizálhatók, ha az olefineket persavakkal reagáltatják.

Az epoxidok előállításának másik módszere ugyancsak olefinekből indul ki. A kettőskötésre először hipoklórossavat addícionálunk, majd a képződött klórhidrint lúggal reagáltatjuk.
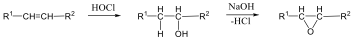
Fizikai tulajdonságok
A dimetil-éter gázhalmazállapotú, a dietil-éter és a kis szénatomszámú éterek folyadékok, míg a 18-nál több szénatomot tartalmazók szobahőmérsékleten kristályosak. Forráspontjuk minden esetben sokkal alacsonyabb, mint az azonos molekulatömegű alkoholoké, mivel az éter molekulák nem asszociálódnak. Apolárisak, vízben alig, vagy nagyon rosszul, de szerves oldószerekben jól oldódnak. Kitűnően oldják a zsírokat.
Kémiai tulajdonságok
Az éterek kémiai tulajdonságait az éterkötésben lévő oxigénatom nukleofil jellege és a különböző vegyületekben a C-O-C kötésszög határozza meg. A 3- és 4-tagú gyűrűs éterekben ez a szögérték jelentősen eltér pl. a nyílt láncú származékokban kb 110°–os értéktől, az epoxidokban ez 60°, ami jelentős feszültséget okoz a gyűrűben. Ezért az ilyen feszült gyűrűs éterek meglehetősen nagy reakciókészséget mutatnak, könnyen felnyílik a gyűrű. Az éterkötésben lévő oxigénatom nemkötő elektronpárja miatti nukleofil jelleg függ attól, hogy az oxigén milyen szénatomhoz kapcsolódik. Ez a nukleofil jelleg azonban az étereket általában a bázisokkal szemben ellenállóvá teszi. Kivételek itt is az epoxidok, amelyek vizes, lúgos közegben 1,2-diolokká,
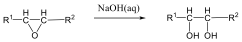
Ammóniával pedig 1,2-aminoalkoholokká alakulnak át.
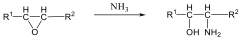
Az epoxidok reagálnak alkoholokkal is, akár lúg, akár savkatalízissel. A reakció vicinális diolok féléteréhez vezet.
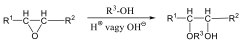
Ahol az éterkötés nem feszült gyűrűs rendszerven található, tehát a C-O-C kötés megközelíti a tetraéderes vegyértékszöget, azokban a vegyületek meglehetősen stabilak, nehezen bonthatók meg és vihetők reakcióba. Lúgoknak, híg savaknak, oxidálószereknek, fémnátriumnak ellenállnak. Erőteljes oxidatív behatással lehet felszakítani az ilyen vegyületekben lévő éterkötést, a reakció ekkor karbonsavakat eredményez.
Az éterek savasan hasíthatók, a folyamatot acidolízisnek nevezzük. Ezt legkönnyebben tömény hidrogén-jodid oldattal lehet melegítés közben kiváltani, de magasabb hőmérsékleten, nyomás alatt sósavval és hidrogén-bromiddal is lejátszódik a reakció. Ha vegyes éterből indultunk ki, akkor általában a kisebb csoportból képződik az alkil-halogenid (R2<R1).

Metil- és etiléterek esetében a fenti reakció mennyiségi meghatározásra is alkalmazható, mert a képződő metil-jodid, illetve etil-jodid jégecetes brómoldatba desztillálva ekvivalens mennyiségben jódsavvá alakul, ami jodometriásan mérhető és a metoxi és etoxicsoportok mennyisége meghatározható.
Az éterekben lévő oxigénnek a kötésben részt nem vevő elektronpárja elektronhiányos molekulákkal koordinatív kötést hozhat létre. Ezekben a komplexekben az éter elektron donorként vesz részt. A legfontosabb ilyen származékok a fémorganikus vegyületek között tárgyalt Grignard-vegyületek, de ide tartoznak a bór-trifluorid éterekkel alkotott ún. bór-trifluorid-éterátok is, illetve az erős ásványi savakkal létrejövő oxóniumsók.
 |
 | |
|
bór-trifluorid-éterát |
oxóniumsó |
Az enoléterek könnyen elhasíthatók savas közegben, amikor alkohol és oxovegyület keletkezik.

Fontosabb éterek és felhasználásuk
Dietil-éter. A legismertebb, legfontosabb, röviden csak éterként nevezett vegyület. Előállítása etanolból vízelvonással, vagy etilénből történik. Színtelen, alacsony forráspontú, jellegzetes szagú folyadék, amelynek kezelése nagy elővigyázatosságot követel meg, ugyanis rendkívül gyúlékony. Állás közben, mint minden éterben, peroxidok képződnek benne, így óvatosság szükséges a desztillálásakor is. Vízzel nem, alkoholokkal viszont elegyedik, jól oldja a zsírokat, de általában jó oldószer, ezért használják főleg laboratóriumi méretben. Régebben altatásra használták, de toxikus hatása miatt ma már nem alkalmazzák.
Etilén-oxid az epoxidok legfontosabb képviselője. Szobahőmérsékleten gáz, forráspontja 11-12 °C, tűz- és robbanásveszélyes anyag. Szaga a dietil-éteréhez hasonló, Előállítása etilénből katalitikus oxidációval, vagy az előzőekben ismertetett hipoklórossavval nyerhető klórhidrin lúgos közegben végrehajtott átalakításával történik. A vegyiparban nagyon fontos alapanyag, nagy mennyiségben használják etilénglikol, dietilénglikol, etanolamin, dioxán stb. előállítására. Orvosi műszerek sterilezésére is használják, de rendkívüli toxikussága, karcinogenitása miatt több helyen már betiltották.
 |
etilén-oxid |
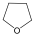 |
tetrahidrofurán |
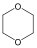 | dioxán |
Tetrahidrofurán, röviden THF, színtelen, jellegzetes szagú folyadék. Előállítása bután-1,4-diolból történik. Mérgező tulajdonságú. Fontos oldószer és alapanyag a vegyiparban.
A dioxán szobahőmérsékleten folyadék, kellemes szaga van. Előállítása etilén-oxid dimerizálásával, vagy etilénglikol dehidratálásával történik. Sok anyagnak kitűnő oldószere. Érdekessége, hogy vízzel minden arányban elegyedik.
Az etilénglikol-monometil- és monoetil-étere (metilcelloszolv, ill. celloszolv) a lakkiparban használatosak, vízzel minden arányban elegyednek. Előállításuk etilén-oxid és metanol, ill. etanol reakciójával történik.
|
|
| |
|
etilénglikol-monometil-éter |
etilénglikol-monoetil-éter |
A dietilénglikol magas forráspontú monoétereit, vagy ezek elegyét gépkocsikban fékfolyadéknak, ill. hidraulikus gépekben használják.
Feladatok
-
Hogyan csoportosíthatók az alkoholok?
-
Melyek az alkoholok jellemző fizikai tulajdonságai?
-
Milyen kiindulási vegyületekből és milyen reakcióval állíthatók elő egyértékű alkoholok?
-
Hogyan tud előállítani Grignard-reagens felhasználásával n-butanolt?
-
Milyen kiindulási vegyületből és milyen reakcióval tud izopropil-alkoholt előállítani?
-
Melyek az egyértékű alkoholok legjellemzőbb kémiai tulajdonságai?
-
Melyek a legismertebb egyértékű alkoholok, mire használják fel őket?
-
Írja fel a négy izomer butil-alkohol szerkezeti képletét és adja meg, hogy melyik adja a jodoform próbát?
-
Milyen módszerek ismeretesek többértékű alkoholok előállítására?
-
Melyek a többértékű alkoholok jellemző reakciói?
-
Milyen ipari módszereket ismer a fenol előállítására?
-
Hasonlítsa össze a fenolok és az alkoholok savasságát!
-
Melyek a fenolok legfontosabb reakciói?
-
Írja fel az alábbi vegyületek szerkezeti képletét: rezorcin, fenil-metil-éter, 2-naftol, propán-1,2-diol, hidrokinon.
-
Hogyan tud etil-n-butil-étert előállítani?
-
Milyen vegyületekké tudja az epoxidokat átalakítani?
-
Nevezze el az alábbi vegyületeket!
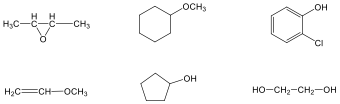
másik termék pedig a glicerin. másik termék pedig a glicerin.
Tartalom
- 14.
- Tiolok, diszulfidok
- Előfordulásuk
- Tiolok nevezéktana
- Tiolok előállítása
- Tiolok fizikai tulajdonságai
- Tiolok kémiai tulajdonságai, reakciói
- Biológiailag fontos tiolok
- Szulfidok (tioéterek)
- Előfordulásuk
- Nevezéktan
- Elektronszerkezet
- Szulfidok előállítása
- Szulfidok fizikai tulajdonságai
- Szulfidok kémiai reakciói
- Szulfidok biológiailag fontos származékai
- Kérdések, feladatok
Tartalom
- Tiolok, diszulfidok
- Előfordulásuk
- Tiolok nevezéktana
- Tiolok előállítása
- Tiolok fizikai tulajdonságai
- Tiolok kémiai tulajdonságai, reakciói
- Biológiailag fontos tiolok
- Szulfidok (tioéterek)
- Előfordulásuk
- Nevezéktan
- Elektronszerkezet
- Szulfidok előállítása
- Szulfidok fizikai tulajdonságai
- Szulfidok kémiai reakciói
- Szulfidok biológiailag fontos származékai
- Kérdések, feladatok
A periódusos rendszerben a kénatom az oxigénnel egy oszlopban, közvetlenül az oxigén alatt helyezkedik el. A külső elektronhéjon mindkét atom esetében 6 elektron található, elektronkonfigurációjuk megegyezik: s2p4. Az oxigén vegyületeiben sp3 hibridállapotú, mint például az alkoholokban vagy az éterekben és sp2 hibridállapotú lehet, mint a karbonilvegyületekben. Az alkoholok kéntartalmú analógjait tioloknak, az éterek kéntartalmú analógjait tioétereknek, a karbonilvegyületek kéntartalmú analógjait pedig tiokarbonil vegyületeknek nevezik.
Míg az oxigénnél ezek az elektronok a 2-es, addig a kénnél a 3-as főkvantumszámú héjon találhatók. A kénatom esetében azonban a 3-as főkvantumszámú héjnak megfelelően d-pályák is vannak, ezért a kénatom hibridizációja ezekre a pályákra is kiterjedhet, a két elem származékainak eltérő fizikai, kémiai tulajdonságai is erre vezethetők vissza.
A tiolcsoport számos, biológiai rendszereket felépítő molekulában megtalálható, így a cisztein nevű aminosavban és a koenzim-A-ban.
Az SH-csoportot tartalmazó kénvegyületeket –tiol utótaggal ellátva tioloknak nevezzük. Előtagként a szulfanil megnevezést használják.
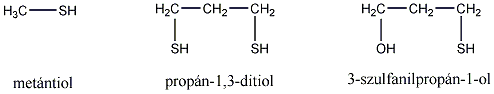
Alkil-halogenidek lúgos közegben kén-hidrogén felesleg mellett tiolokká alakulnak:
![]()
A tiolok, különösen a kis szénatomszámú származékaik átható, kellemetlen szagú vegyületek.
A tioloknak alacsonyabb a forráspontja, mint az azonos szénatomszámú alkoholoknak. Mivel a kénatom elektronegativitása a szénatoméval megegyezik és ez az érték a hidrogénatom elektronegativitásától is alig tér el, az S-H kötés gyakorlatilag apoláris. A tiolokban a kénatomnak ugyan van két nemkötő elektronpárja, de az S-H kötés apoláris jellege miatt a kénatom nem tud protont koordinálni, ezért a tiolok hidrogénkötéssel nem asszociálódnak. Ez magyarázza az alacsony forráspontot:

Ugyan a tiolok apoláris vegyületek, ennek ellenére mégis erősebb savak, mint az alkoholok. A nagyobb méretű kénatom könnyebben polarizálható, mint az oxigén, ezért az S-H kötés gyengébb, mint az O-H kötés. Emellett a kénnek az oxigénnél nagyobb elektronhéján az elektronok nagyobb térrészen oszlanak meg, könnyebben elviselve a protonleadás után megjelenő negatív töltést.
![]()
A gyengébb S-H kötéssel függ össze a tiolok nagyfokú érzékenysége oxidálószerekkel szemben. Már a levegő oxigénjének vagy más enyhe oxidálószernek a jelenléte is diszulfidokhoz vezet. A reakció reverzibilis, így redukálószerek hatására visszanyerhető a tiol.
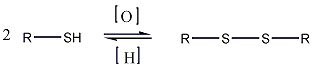
Erélyes oxidálószerek hatására (KMnO4) a tiolok szulfinsavakká illetve szulfonsavakká oxidálódnak:
![]()
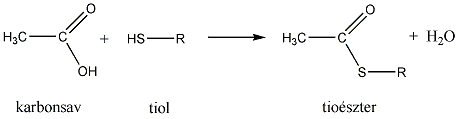
A cisztein aminosav oxidációjával kialakuló –S-S- diszulfidhidak hozzájárulnak a fehérjék másodlagos szerkezetének kialakításához.
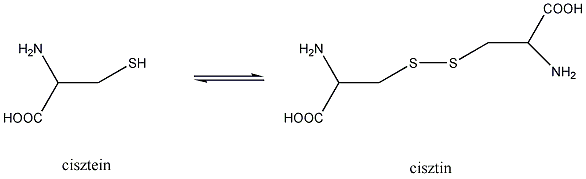
Jelentős mennyiségben található a sejtekben a glutation (γ-glutamil-ciszteinil-glicin) nevű tripeptid. Két glutation tiolcsoportja oxidáció hatására diszulfidkötéssel kapcsolódik össze és oxidált glutation képződik. A redukált és oxidált glutation a sejtekben redoxirendszerként működik.
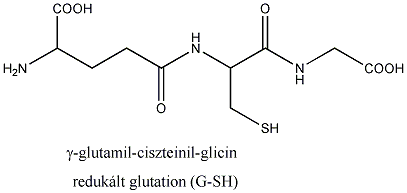
Az α-liponsav öttagú, telített gyűrűje két kénatomot tartalmaz, a piruvát-dehidrogenáz enzimkomplexben koenzimként működik. Az -S-S- kötés reverzibilisen redukálódik és α-dihidroliponsav keletkezik:
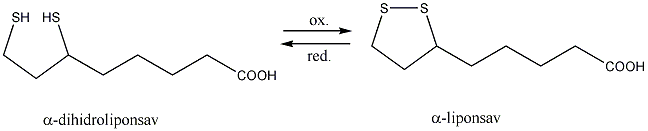
A koenzim-A-ból keletkező tioészter, az acetil-koenzim-A fontos szerepet játszik a zsírsavak és az izoprénvázas vegyületek bioszintézisében:
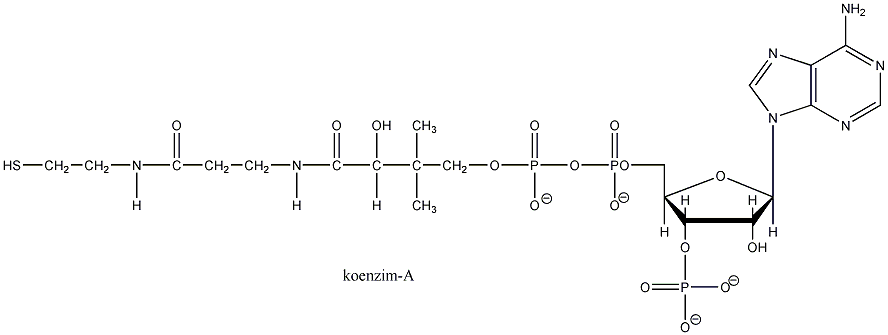
A szulfidok is gyakran előfordulnak biomolekulákban, pl. aminosavban (metionin), vitaminban (biotin), antibiotikumokban (penicillinek, kefalosporinok).
Leggyakrabban a csoportfunkciós elnevezést alkalmazzák, melyben a szulfid funkciós csoportnév előtt betűrendben, kötőjellel elválasztva az alkilcsoportok nevét soroljuk fel:
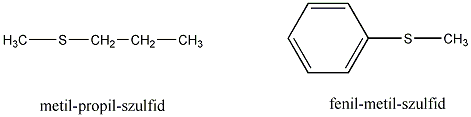
A C-S kötéshossz jóval nagyobb, mint a C-O kötéshossz, ennek megfelelően a kötési energiák a kötéshosszal fordítottan arányosak, a C-S kötés energiája kisebb.
Az éterek és a tioéterek kötésszögében is eltérés mutatkozik, azonos szubsztituensek esetén a kénvegyületekben a kötésszögek jóval kisebbek. Míg pl. a dimetil-éterben a C-O-C kötésszög 112 º, addig a dimetil-szulfidban csak 98,5 º.
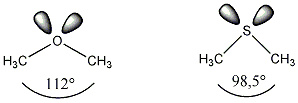
A tiolalkoholok nátrium-hidroxiddal tiolátokká alakíthatók, majd alkil-halogenidekkel szulfidokká alkilezhetők:
![]()
Az illékony tioéterek a tiolokhoz hasonlóan jellegzetes, kellemetlen szagú vegyületek.
Az analóg éterekhez képest valamelyest magasabb olvadás- és forrásponttal rendelkeznek, valamint vízben kevésbé oldódnak.
A tioéterek könnyen oxidálhatók, enyhe körülmények között szulfoxidokká, erélyesebb oxidálószerek hatására szulfonokká alakíthatók:
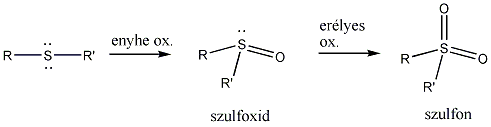
A kén és az oxigén eltérő elektronszerkezetéből adódóan a kénatom nukleofilitása lényegesen nagyobb az oxigénatoménál, ezért a tioéterek az éterektől eltérően készségesen reagálnak alkil-halogenidekkel szulfóniumsókat képezve:
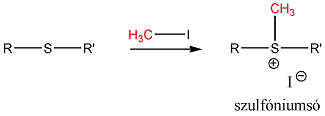
A szulfónium sókban a kénatomhoz kapcsolódó szénatomok pozitív polározottsága megnő, ezért ezek a vegyületek alkilezőszerekként is használhatók:
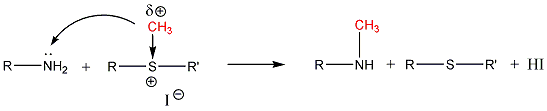
Az élő szervezetben lejátszódó biológiai metilezés is a trialkilszulfónium sóknak ezen a tulajdonságán alapszik. A metionin nevű aminosav, amely egy vegyes szulfidnak tekinthető, a trifoszfátcsoport lehasadása közben reagál az ATP-vel, S-adenozil-metionin (SAM) keletkezik.
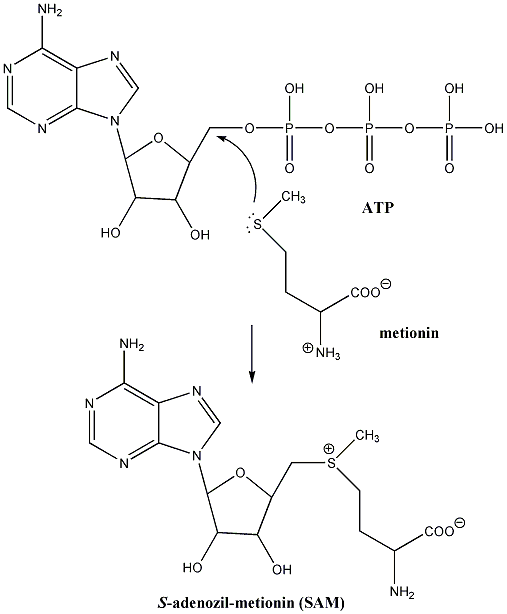
A trialkilszulfónium só szerkezetű SAM szolgáltatja a metilcsoportot a biológiai nukleofilek számára, így keletkezik például a nornikotinból nikotin, a noradrenalinból adrenalin és az etanolaminból kolin.
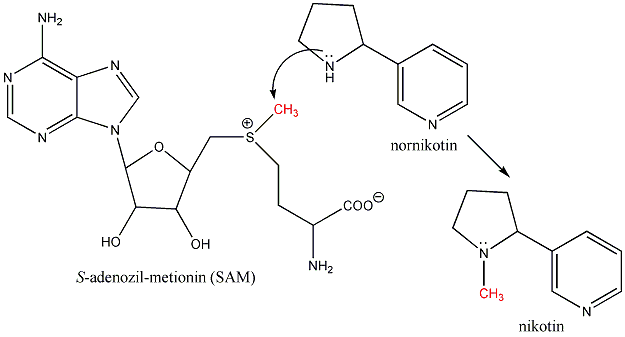
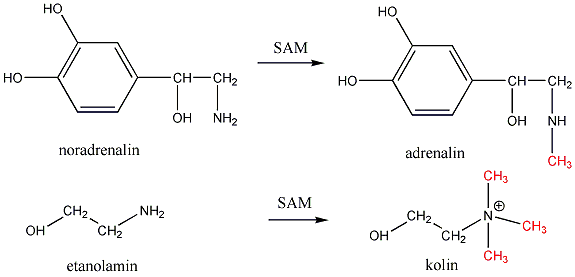
Telített tiazolvázat, tiazolidingyűrűt találunk a két legismertebb antibiotikumcsalád, a penicillinek és a kefalosporinok alapvázában. Bioszintézisük során a penicillinek és kefalosporinok váza egy kéntartalmú aminosavból – a ciszteinből – és valinból épül fel:
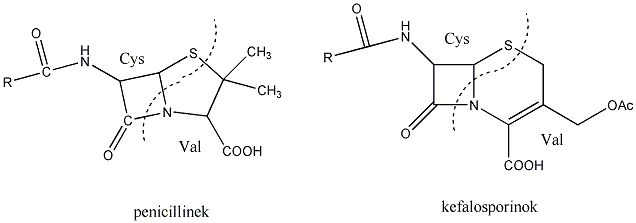
Az 1930-as években Gerhard Domagk ismerte fel, hogy a prontozil márkanevű azofesték bizonyos Gram-pozitív baktériumokat elpusztít. A vizsgálatok során kiderült, hogy a hatás hordozója nem maga a festék, hanem annak sejten belüli bomlásakor keletkező p-amino-benzol-szulfonamid. A szulfanilamid szulfonamido-csoportjának egyik hidrogénjének kicserélésével számos szulfonamid-származékot állítottak elő:
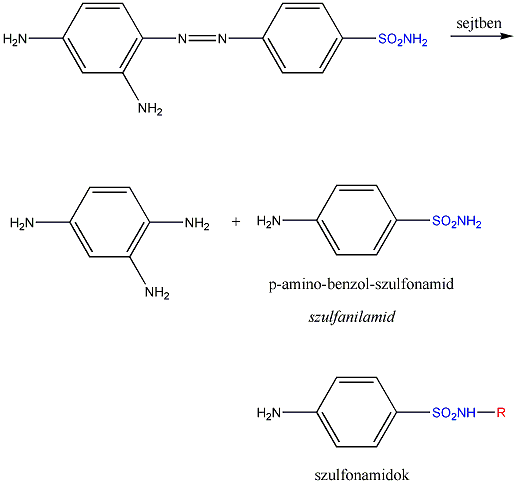
A hatásmechanizmus lényege, hogy a szulfonamid szerkezetileg hasonló a p-amino-benzoesav (PABA) molekulájához, ezért gátolja a PABA beépülését a baktériumok növekedéséhez feltétlenül szükséges folsav szintézisénél.
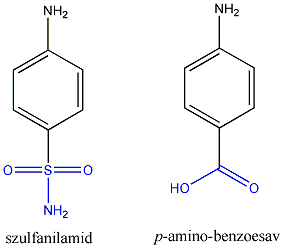
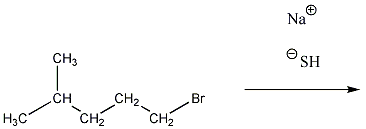
Tartalom
A nitrovegyületek lehetnek egy- és többértékűek, ill. attól függően, hogy a nitrocsoport hányad rendű szénatomhoz kapcsolódik, beszélhetünk első-, másod- és harmadrendű nitrovegyületekről.
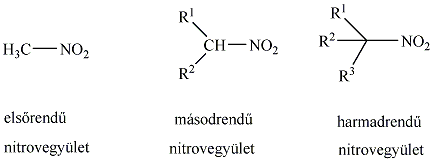
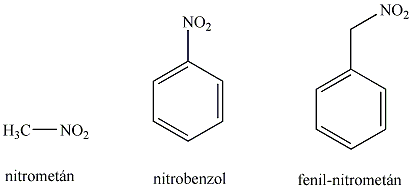
A nitrocsoport az alábbi két határszerkezettel (A és B) írható le, illetve a delokalizáció a C szerkezettel is kifejezhető.
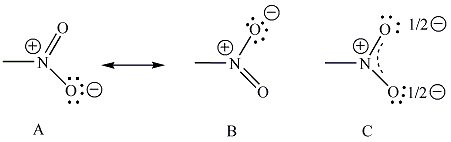
A 4π-elektronos konjugált elektronrendszerhez az oxigének 1-1, míg a nitrogén 2 vegyértékelektronjával „járul” hozzá, ugyanakkor az oxigének megtartják két nem kötő párjukat, az oxigéneken a negatív töltés megoszlik, a nitrogénen egységnyi pozitív töltés van.
Alkánok salétromsavas nitrálásával (főleg iparban):
![]()
Alkil-halogenid és nátrium-nitrit reakciójával (laborban):
![]()
A reakciókörülményektől függően a nitrit anion ambidens jellege miatt (az oxigén és a nitrogén is lehet nukleofil) az alifás nitrovegyület mellett salétromossav észter is képződhet.
Aromás nitrovegyületek előállítása kénsav és salétromsav elegyével (nitrálósav) aromás elektrofil szubsztitúcióval történik, ahol a nitróniumkation NO2+ a nitráló ágens:
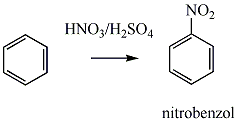
A nitrovegyületek között fellépő dipól-dipól kölcsönhatás miatt magas olvadás és forrásponttal rendelkeznek. A nitroparaffinok színtelen, jellegzetes szagú vegyületek. Vízben nem, lúgban jól oldódnak, szerves oldószerekkel elegyednek. A mononitro származékok színtelenek, a polinitroszármazékok sárgák.
A nitrovegyületek kémiai sajátságait a nitrocsoport erős elektronvonzó sajátsága határozza meg. A primer és a szekunder alifás nitrovegyületek a CH-savak körébe tartoznak, bázisokkal sót képeznek. Az alifás nitrovegyület deprotonálódásakor mezomerstabilizált nitronát-anion képződik, melynek protonálása mind a nitroformát, mind az aciformát eredményezheti, a nitroforma termodinamikailag stabilisabb, viszont az aciforma képződik gyorsabban.
A nitrovegyületek bázis jelenlétében aldehidekkel nitroaldol kondenzációs reakcióba lépnek, pl. nitrometánnal benzaldehidből β-telítetlen nitrovegyület, β-nitrosztirol állítható elő.
Az aromás vegyületek nitrocsoportja savas közegben és lúgos közegben is aminná redukálható. Például a nitrobenzol redukálásával anilinhez jutunk. Lúgos közeg esetén több vegyület is elkülöníthető:
Az aromás nitrovegyületek aromás elektrofil szubsztítúciós reakcióban a nitrocsoport – I és –M effektusa miatt kevésbé reakcióképesek. Pl. a nitrobenzolt nitrálva jóval erélyesebb körülmények között jutunk 1,3-dinitrobenzolhoz, mint a benzol nitrálásánál alkalmazott körülmények.
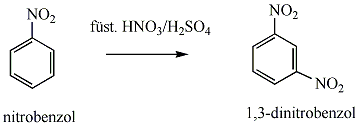
A halogénezett polinitrovegyületek halogénatomja aromás nukleofil szubsztitúciós reakcióban cserélhető, pl. a 2,4-dinitro-fluorbenzol peptidkémiai reagens, amely aminosavak aminocsoportjával már enyhe melegítéskor reagál vizes, alkoholos NaHCO3 jelenlétében
A negyvenes évek második felében izolálták a Streptomyces venezuelae baktérium által termelt szélesspektrumú antibiotikumot a klóramfenikolt, azonban ma már toxicitása miatt kevésbé használatos. Bár nem nitrovegyület (mert nem tartalmaz C-NO2 kötést), mégis itt említjük a glicerin trinitrátot (helytelenül nitroglicerin), amely a glicerin salétromsavas észtere. A robbanóanyag iparban is alkalmazzák és a gyógyászatban pedig értágítóként használatos. Utóbbi területen szintén megjelentek korszerűbb készítmények.
Tartalom
Az aminok feloszthatók rendűségük és értékűségük alapján, illetve az alapján is, hogy az aminocsoport milyen szénlánchoz kapcsolódik. A rendűség alatt azt értjük, hogy a nitrogénatomhoz hány alifás vagy aromás szénhidrogéncsoport kapcsolódik, így beszélhetünk primer, szekunder, tercier aminokról ill. kvaterner ammóniumvegyületekről:
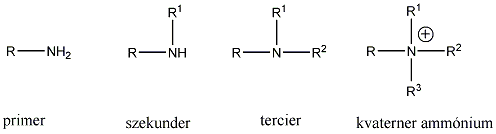
Értékűség szerint lehetnek egyértékűek pl.: metilamin, kétértékűek pl.: etiléndiamin, vagy poliaminok, pl. hexametiléntetraamin.
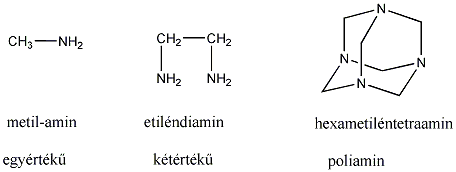
Az aminok elnevezése háromféleképpen lehetséges:
*Csoportfunkciós név: az alapszénhidrogént funkciós csoportként nevezzük el és “amin” utótaggal látjuk el
**Szubsztitúciós név: bonyolultabb aminok estében használatos, alapszénhidrogén neve + amin, diamin, triamin végződés
Triviális név: főleg nitrogéntartalmú heterociklusoknál használják, de néha elfogadott mind az alifás mind az aromás aminok jelölésére. ***
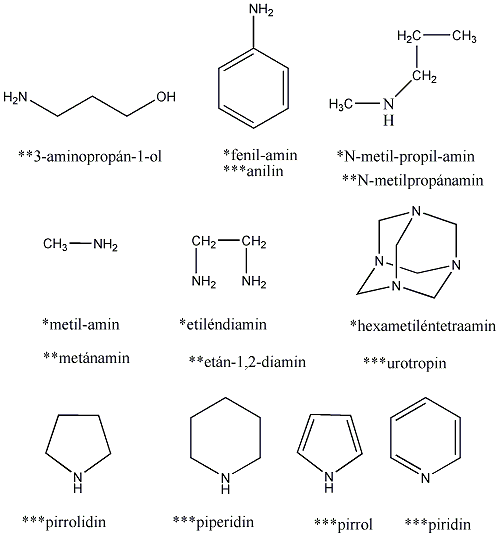
Az aminok szerkezete a nitrogénatom sp3 hibridizációja miatt trigonális piramisos, az N-C vegyértékszögek 108º-ot zárnak be. Ha a nitrogénatom nemkötő elektronpárját ligandumnak tekintjük, akkor tetraéderes elrendeződésről beszélhetünk, és így a különböző szubsztituenseket tartalmazó tercier aminok szimmetriatulajdonságai alapján királis vegyületeknek tekinthetők. Azonban a nitrogénatomhoz kapcsolódó ligandumok térhelyzetüket változtatják, a N-atom az alapon (a ligandumjai által meghatározott síkon) keresztül nagyon könnyen átrezeg, és a piramis kifordul. Ezt a folyamatot konfigurációs vagy piramidális inverziónak nevezzük.
A folyamat energia igénye kicsi (20-25 kJ/mol), de lényegesen nagyobb lehet ciklusos származékoknál, például aziridinekben (77 kJ/mol). Így pl. az alábbi aziridin-származékok elválaszthatók:
Aminokat előállíthatunk az ammónia alkilezésével. Első lépésben primer ammóniumsó képződik, amelyet a feleslegben levő ammónia deprotonál és primer amin képződik, a folyamat azonban tovább megy, szekunder amin, tercier amin és végül kvaterner ammóniumsó képződik. Ha az alkilezőszer van feleslegben, akkor kvaterner ammóniumsó lesz a főtermék, ha az ammónia, akkor elsősorban primer amin képződik:
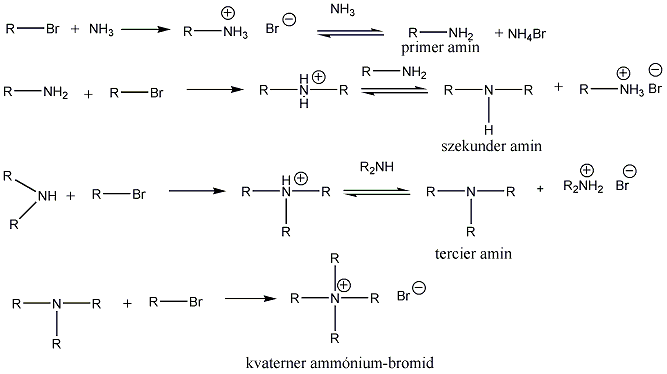
Gabriel-szintézissel primer aminokat nyerhetünk. A ftálimid káliumsóját alkilezve, majd a kapott N-alkil-ftálimidet hidrazinnal reagáltatva felszabadul a primer amin:
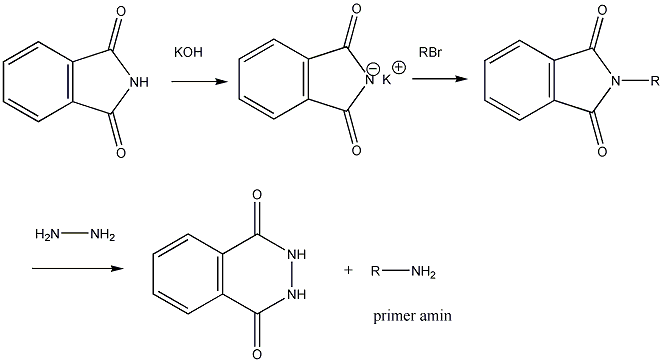
Aromás aminokat nitrovegyületek redukciójával tudunk előállítani vas és sósav jelenlétében:
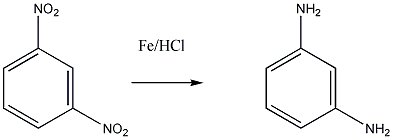
Alkil-halogenidekből nyerhető nitrileket, illetve azidokat lítium-alumínium-hidriddel redukálva primer aminokat kapunk:
![]()
Aldehidből ill. ketonból ammóniával vagy primer aminnal aldimin ill. ketimin képződik (Schiff-bázis) amely katalitikus hidrogénezéssel vagy komplex fémhidrides redukcióval aminná alakítható:
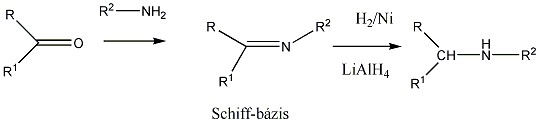
Aminok előállítása lebontással: savamidok lúg feleslegben, hipobromit oldattal kezelve eggyel rövidebb szénláncú primer amint adnak (Hofmann-féle lebontás):
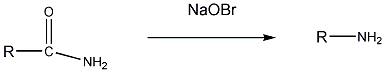
Az aminoknak sokkal magasabb a forráspontjuk, mint a hasonló molekulatömegű szénhidrogéneknek. A kis szénatomszámú aminok dipól-dipól kölcsönhatásba lépnek, ill hidrogénkötéseket képeznek. Az intermolekuláris hidrogénhíd-kötődések az aminokban gyengébbek, mint az alkoholokban. Így a metil-amin szobahőmérsékleten gáz halmazállapotú, míg a metanol folyadék. A kis szénatomszámú aminok vízben jól oldódnak, a nagyobb szénatomszámú aminok és az aromás aminok a vízoldékonysága kisebb. Az aminok sói és a kvaterner ammónium vegyületek vízodhatóak. Az aminok kellemetlen szagú vegyületek.
Az aminok bázicitása a közeg függvényében a rendűségtől függ. Gázfázisban a bázicitást a szubsztituensek elektromos hatása határozza meg, elektronküldő csoportok növelik a nitrogénatom elektronsűrűségét és így a bázicitást is. A bázicitás sorrendje gázfázisban:
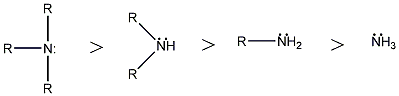
Vizes közegben az aminok bázicitását végső soron az határozza meg, hogy az alábbi egyensúlyi folyamat (a nitrogén protonálódása és a hidroxidionok keletkezése) mennyire eltolódik el a felső vagy az alsó nyíl irányába.
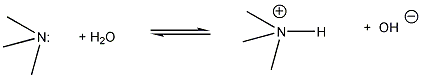
A vizes közegben az alkilcsoportnak kettős hatása van az ammóniumionra: egyrészt elektronküldő sajátsága révén stabilizálja azt, másrészt viszont nehezíti a szolvatációt.
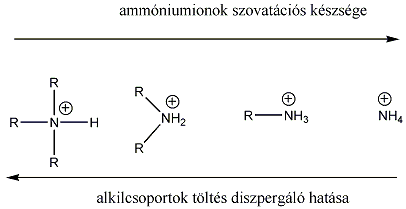
A két hatás eredője így az lesz, hogy a szekunder aminok a legbázisosabbak, őket követik a primer aminok, tercier aminok, ammónia, majd az anilin.
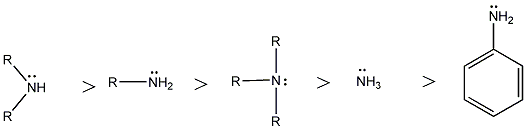
Az aromás aminok sokkal gyengébb bázisok, mint az alifás analógjaik. A nitrogénatom elektronpárja ugyanis az aromás gyűrű pz-pályáival kölcsönhatásba lép, ezért a nitrogénatom nehezebben protonálódik. Az anilin elektronszerkezetét az alábbi ábra mutatja:
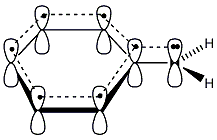
Az aminok acilezésekor a primer vagy szekunder aminok egyik hidrogénjét acilcsoporttal helyettesítjük, és N-mono vagy N,N-diszubsztituált savamidokhoz jutunk (X=halogén vagy O-alkil vagy R3COO).
Az aminokat szulfonsav-kloridokkal reagáltatva szulfonamidok nyerhetők, ezt a reakciót különböző rendű aminok szétválasztására lehet használni (Hinsberg-féle elválasztás). A benzoszulfonsav-klorid hatására a primer aminokból lúgban oldódó szulfonamid keletkezik, a szekunder szulfonamid olaj vagy csapadék, a tercier amin pedig nem képez szulfonamidot.
Primer aminok salétromossavval instabil alkil-diazóniumvegyületet képeznek, amely szobahőmérsékleten bomlékony és így keveréktermék képződhet (alkohol, olefin, alkil halogenid).
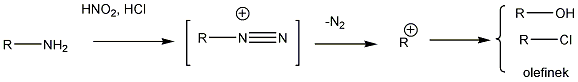 Az aromás primer aminokból képződő diazóniumvegyületek + 5 °C-ig stabilisak, és nitrogén fejlődéssel az új szubsztituens a diazóniumcsoport helyébe lép be. Így aril-jodidokat, kloridokat, cianidokat, fluoridokat lehet előállítani ill. a diazóniumcsoport helyére hidrogén atomot tudunk beépíteni.
Az aromás primer aminokból képződő diazóniumvegyületek + 5 °C-ig stabilisak, és nitrogén fejlődéssel az új szubsztituens a diazóniumcsoport helyébe lép be. Így aril-jodidokat, kloridokat, cianidokat, fluoridokat lehet előállítani ill. a diazóniumcsoport helyére hidrogén atomot tudunk beépíteni.
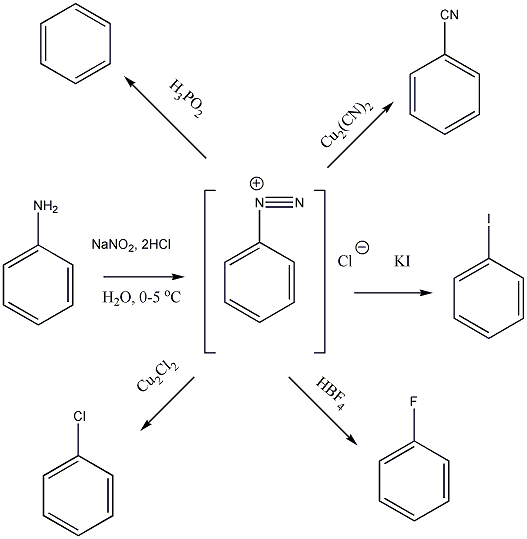
Alifás és aromás szekunder aminokból N-nitrozoaminok képződnek.
Az alifás tercier aminok salétromossavval sót, míg az aromás tercier aminok C-nitrozo vegyületet képeznek.
Az aromás aminok kálium-bikromáttal p-benzokinonná alakulnak. A tercier aminok oxidációja N-oxidokhoz vezet, gyűrűs szekunder aminok pedig nitronokat adnak:
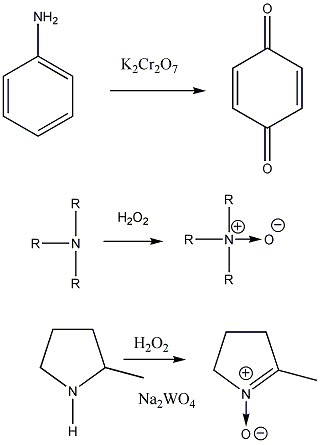
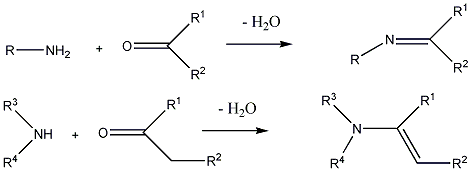
Tartalom
A szervezetben található biológiailag fontos aminok az α-aminosavak dekarboxileződésével keletkeznek. Így ornitinből putreszcin képződik, hisztidinből hisztamin. Utóbbi vegyület fontos szerepet játszik az allergiás folyamatokban.
A szerin aminosavból a 2-aminoetán-1-ol (kolamin) keletkezik, amely biológiai metilezés során kolinná alakul. A kolamint a kefalin tartalmazza, míg a kolin a lecitin molekulájában található meg. A kolint acilezve acetilkolin képződik, amely a paraszimpatikus idegrendszer ingerületátvivő anyaga.
Biológiai szempontból fontos vegyület a dopamin, amely a tirozinból származtatható. A dopamin az idegingerületátvitelben vesz részt. A DOPA-t dopaminszint-növelő szerként Parkinson-kór kezelésére használják A dopamin hidroxileződésével a noradrenalin majd metileződéssel az adrenalin jön létre. Utóbbi a mellékvese által termelt hormon, amely a véráramba jutva vérnyomás emelkedést, a glikogén lebontásának sebességét növelve vércukorszint emelkedést okoz.
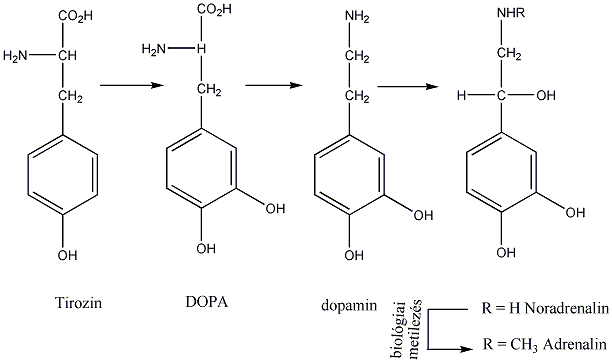
Az amfetamin (régen stimuláló, éhségérzet csökkentésére használt gyógyszer) hírhedt származéka a diszkódrogként használatos szer, az eksztázi. Szerkezetük hasonlatos mind a dopaminhoz, mind a szerotoninhoz.
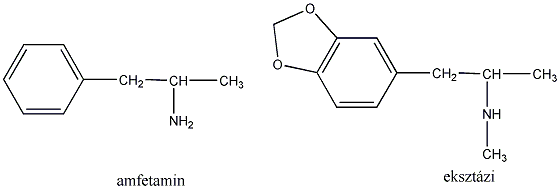
A szerotonin a triptofánból keletkezik, a központi idegrendszer fontos neurotranszmittere.
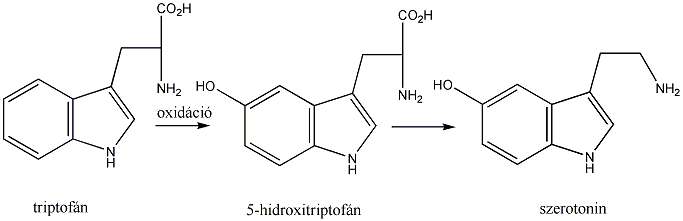
Az alkaloidok olyan nitrogéntartalmú növényi bázisok, amelyeknek pontosan körülírható fiziológiás hatásuk van. Több mint tízezer alkaloid ismert, közülük több vegyületet vagy azok módosított származékait a gyógyászatban is alkalmazzák.
Az alkaloidokat többféleképpen csoportosíthatjuk, így megkülönböztethetünk aminosavból felépülő egyszerű alkaloidokat ún. protoalkaloidokat (pl. piperin, meszkalin), nem aminosav eredetű alkaloidokat (pl. szteroid alkaloidok) ezek a pszeudoalkaloidok. Beszélhetünk ún. valódi alkaloidokról, amelyek aminosavból képződnek, de a nitrogénjük a bioszintézis folyamán heterociklus részévé vált. Az alkaloidokat csoportosíthatjuk eredetük szerint, azaz a növénycsaládok alapján, vagy gyűrűs szerkezetüknek megfelelően. Újabban a bioszintézisük kiinduló vegyülete alapján szokás osztályozni (pl. ornitin, lizin, fenilalanin, tirozin, triptofán, hisztidin, nikotinsav, antranilsav alapú alkaloidok).
A nikotin, a dohány (Nicotiana tabacum) főalkaloidja, nagyon toxikus, magas forrpontú, vízgőzzel illó folyadék. Kis dózisban élénkítő hatású. Az atropin a maszlagos nadragulya (Atropa belladonna) hatóanyaga, ugyancsak toxikus, szemészetben pupillatágítóként alkalmazzák. A kokacserje (Erythroxylon coca) hatóanyaga a kokain, veszélyes kábítószer, de a gyógyászatban is alkalmazzák, mint helyi érzéstelenítőt.
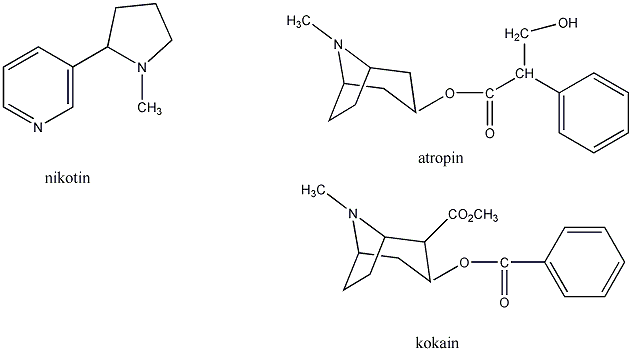
A piperin a feketebors (Piper nigrum) alkaloidja. Már az ókorban is használták a gránátalma (Punica granatum) kivonatát bélférgek hajtására. Ennek a hatóanyaga a pelletierin.
Az efedrin egy ősi gyógynövényben, a csikófarkban (Ephedra vulgaris) fordul elő, és érszűkítő, görcsoldó hatású. Orrcseppekben alkalmazzák. A meszkalin hallucinogén hatású és a Mexikóban honos kaktuszfaj (Anhalonium lewinii) hatóanyaga.
A mákfélék (Papaveraceae) családjába tartozó növényekben a tirozinból többféle vázrendszer alakul ki. Ezek többsége izokinolingyűrűt tartalmaz. A máknövény benzokinolinvázas alkaloidjai közül a természetben a papaverin és származékai mint simaizomgörcsoldók kerülnek alkalmazásra. A máknövény főalkaloidját, a morfint 1930-ig kizárólag ópiumból nyerték. Kabay János magyar gyógyszerésznek köszönhetően a kicsépelt máktok és mákszalma lett e fontos gyógyszer előállításának alapanyaga. A morfin káros mellékhatásai (hozzászokás, eufória) ellenére ma is nélkülözhetetlen fájdalomcsillapító. Az aromás gyűrűn metilezett származéka a kodein, köhögéscsillapító, a diacetil-származéka, a heroin, igen veszélyes kábítószer.
A kinint a kínafa (Cinchona succirubra) kérgéből izolálják és évente kb. 1 millió kilogrammot használnak maláriás betegségek kezelésére. Az anyarozsalkaloidok szintén triptofánból levezethető alkaloidok. Az anyarozs (Secale cornutum) a Claviceps purpurea nevű gomba által okozott fertőzés gabonaszemeken. Az anyarozsból több mint 30 alkaloidot izoláltak, ezek között több sztereoizomer is található. A vegyületek többsége vagy savamid vagy peptid szerkezetű. A vegyületek erélyes lúgos hidrolízisének terméke a lizergsav vagy ennek C-8 epimere, az izolizergsav. Az anyarozs alkaloidok közül csak a lizergsavszármazékok mutatnak élettani hatást. Ezek méhizomzat összehúzó hatásúak, így a szülések utáni vérzés csillapítására használják. Az LSD szintetikus termék, erősen hallucinogén hatású.
1.) Nevezze el az alábbi vegyületeket és adja meg az aminok rendűségét is!
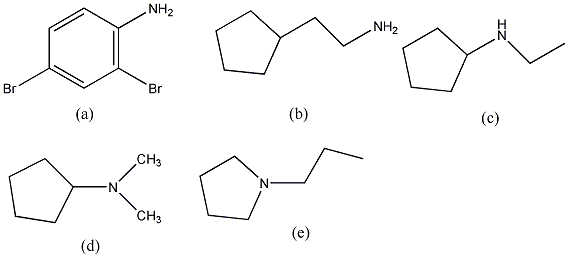
2.) Hogyan szintetizálna anilint?
(a) benzolból (b) toluolból (c) benzamidból
3.) Az alábbi szintézisek helytelenek. Javítsa ki őket!
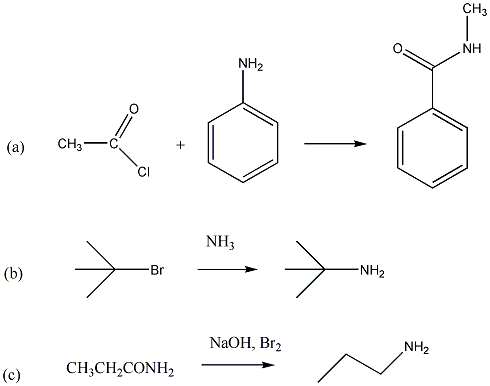
4.) Az alábbi aminok közül melyik bázisosabb?
5.) A Cavinton agyi vérkeringést javító szerként került forgalomba és a hazai gyógyszeripar legsikeresebb eredeti készítménye. Hány aszimmetria centrum van a Cavintonban és melyik a bázisosabb nitrogén?
Tartalom
Tartalom
Az oxovegyületekben egy oxigénatom kettős kötéssel kapcsolódik egy szénatomhoz. A szén- és oxigénatomot együttesen karbonilcsoportnak nevezzük. Amennyiben a karbonilcsoport a lánc végén helyezkedik el aldehidről, amikor pedig a lánc közben ketonról van szó.
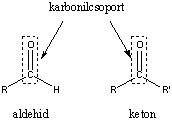
A kisebb szénatomszámú alifás oxovegyületek a természetben nem fordulnak elő, de más funkciós csoportokat is tartalmazó biomolekulákban, pl. szénhidrátokban (polihidroxi-oxovegyületek), ketontestekben (oxo-karbonsavak) gyakoriak.
Az aldehidek szisztematikus neve –al végződést kap, a ketonoké –on végződést, azonban számos vegyület triviális neve az elterjedt:
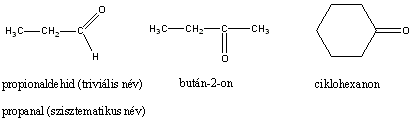
Az sp2 hibridállapotú szén és oxigénatom, valamint a karbonilcsoporthoz kapcsolódó másik két atom egy síkban helyezkednek el. Az karbonilcsoportban az oxigénatom jóval nagyobb elektronegativitása miatt a -kötés elektronpárja az oxigénatom felé tolódik el, polarizált kötés jön létre, dipólusmomentum alakul ki.
Primer alkoholok oxidációjával aldehidek, szekunder alkoholok oxidációjával ketonok keletkeznek:
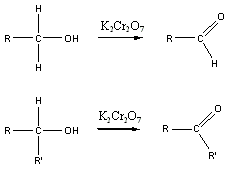
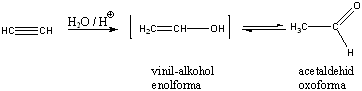
Az oxovegyületek molekulái között dipól-dipól kölcsönhatás jön létre, hidrogénkötést egymás között ezek a molekulák nem tudnak kialakítani. Ezért az oxovegyületek olvadás és forráspontja magasabb, mint a hasonló molekulatömegű paraffinoké, viszont alacsonyabb, mint a hasonló molekulatömegű alkoholoké.
A kis szénatomszámú aldehidek és ketonok vízben jól oldódnak, az oxocsoport a vízzel erős hidrogénkötést alakít ki:
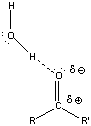
A szénatomszám növekedésével nő az apoláris jelleg, csökken a vízoldhatóság.
Az aldehidek és ketonok reakciókészségét az erősen polározott karbonilcsoport határozza meg. A szén-oxigén kettős kötés pozitívan polározott szénatomja készségesen reagál nukleofil ágensekkel, majd a negatív töltésű oxigénatom protont képes megkötni:
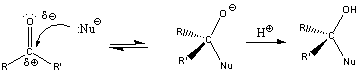
Az oxovegyületek közül az aldehidek vizes oldatban hidrát formában is megtalálhatók, keletkezésük egyensúlyra vezető kémiai reakció.
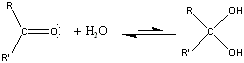
A keletkezett geminális diolok bomlékony vegyületek, csak vizes oldatban léteznek.
Az oxovegyületek alkoholokat is addícionálnak és az aldehidekből először félacetálok, majd acetálok képződnek, a vízaddícióhoz hasonlóan egyensúlyi folyamatban. A félacetál forma csak gyűrűs szerkezetben stabilis (pl. szénhidrátok.) A félacetálok alkoholfelesleg alkalmazása mellett savkatalizált reakcióban acetállá alakulnak át:
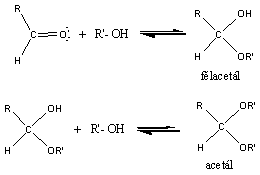
A hidrogén-cianidból lúgos közegben keletkező cianid-anion, mint jó nukleofil ágens, készségesen addícionálódik a karbonilcsoportra, miközben ciánhidrin képződik. A ciánhidrinek sokrétű felhasználást nyerhetnek a szerves kémiai szintézisekben, mivel mind az –OH, mind a –CN csoportjuk változatos módon továbbalakítható. Erre látunk egy jellemző példát az aminosavak Strecker-szintézisénél.
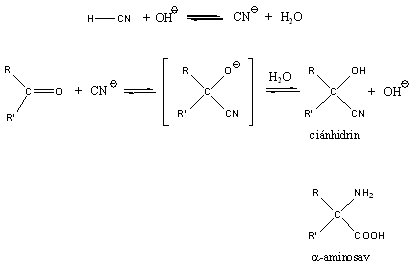
A kondenzációs reakciók többlépéses, addíciós és eliminációs kulcslépésekből álló folyamatok. Az ammónia és származékai az oxovegyületekkel nukleofil addíciós reakcióban reagálnak, majd ez a köztitermék vízmolekula kihasadása közben, eliminációval stabilizálódik és imin, vagy Schiff-bázis keletkezik:
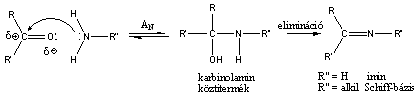
A Schiff-bázis képződés számos biológiai folyamat alapját képezi:
- aldehidek megkötése az enzimfehérje primer aminocsoportot tartalmazó oldalláncával (lizin-oldallánc), pl. a látás folyamatában, amikor a retinal az opszinhoz kapcsolódik és rodopszin keletkezik (ld. Izoprénvázas vegyületek)
- aminosavak és ketosavak transzaminálási reakciója (ld. Aminosavak fejezet)
Az oxovegyületek redukciója alkoholokhoz, oxidációja karbonsavakhoz vezet. A ketonok csak erélyes körülmények között oxidálhatók, oxidációjuk lánchasadás közben két karbonsavat eredményez.
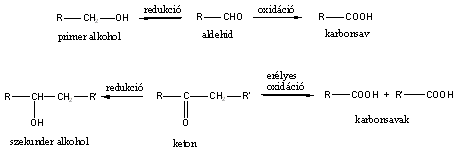
Az aldehidek könnyen oxidálhatók, közben fémionokat redukálnak. Kimutatásuk Fehling- vagy Tollens-próbával is ezen alapul. A Fehling-próba során vörös színű réz(I)-oxid csapadék képződik, a Tollens-próba során pedig az Ag+-ion fémezüstté redukálódik:
Fehling-reakció:
![]()
Tollens-reakció:
![]()
A karbonilcsoport melletti, -szénatomhoz kapcsolódó hidrogénatom a karbonilcsoport elektronszívó (-I effektusa) miatt lazítottá válik, könnyen disszociál:
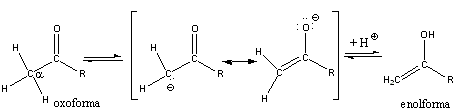
Az egyensúlyi elegyben általában az oxoforma van túlsúlyban, kivéve az olyan vegyületeket, ahol például konjugáció vagy hidrogénkötés stabilizálja az enolformát:
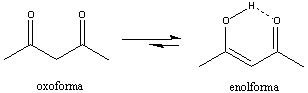
Számos biokémia átalakulás enol-oxo tautomérián keresztül játszódik le:
a., A glikolízis során keletkező három szénatomos szénhidrát molekulák, a D-glicerinaldehid-3-foszfát és a dihidroxi-aceton-foszfát az endiol formán keresztül egyensúlyi folyamatban alakulnak egymásba:
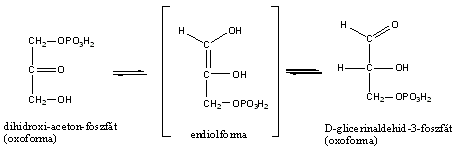
b., Az enol-oxo tautoméria nem csak az α-helyzetű szénatom hidrogénjének vándorlását, hanem α-helyzetű nitrogénatomhoz kötött hidrogén mozgását is jelentheti. Ebben az esetben a tautomer formákra a laktám-laktim elnevezést használják. Ilyen tautomer átalakulások a purin- és pirimidinbázisoknál gyakoriak.
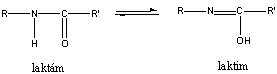
A karbonilvegyületek α-szénatomjáról lúgos közegben egy proton könnyen leszakítható, így karbanion képződik (ld. enol-oxo tautoméria). Ez az anion egy másik aldehidmolekulával szemben nukleofil ágensként viselkedik és nukleofil addíciós reakcióban annak karbonilszénatomjához kapcsolódik. Az alkoxidion a vizes közegben protonálódik, aldollá alakul.
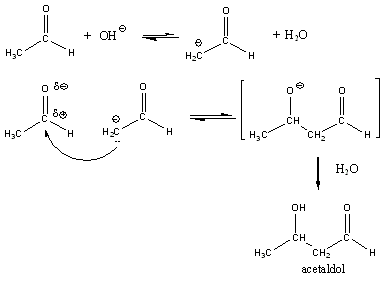
Számos biokémiai folyamat aldolkondenzációval játszódik le, így pl. a fruktóz-1,6-difoszfát keletkezése dihidroxi-aceton-foszfátból és D-glicerinaldehid-foszfátból szintén aldoladdíció:
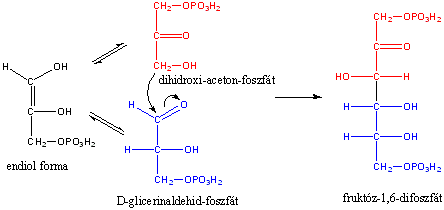
A formaldehid szúrós szagú gáz. Vízben jól oldódik, 35-40 %-os vizes oldata a formalin. A fehérjéket oldatukból kicsapja, baktérium- és gombaölő hatású. A fehérjékben lévő szabad aminocsoporttal készségesen reagál Schiff-bázis képződése közben:
Az acetaldehid alacsony forráspontú (20-21 °C) kicsit savanykás illatú vegyület. A szerves vegyipar egyik alapvegyülete, ecetsavat, észtereket, butadiént állítanak elő belőle.
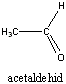
Az aceton alacsony forráspontú (56 °C), kellemes illatú folyadék. Iparban és laboratóriumban elterjedten alkalmazzák oldószerként. Biokémiai folyamatokban a ketontestek közé sorolják, amelyek a májban képződnek és fontos energiaszolgáltató anyagok szénhidrátban szegény étrend vagy éhezés során.
A kinonok telítetlen gyűrűs, konjugált kötésrendszert tartalmazó diketonok. Legfontosabbak az 1,4-benzokinon és az 1,4-naftokinon:
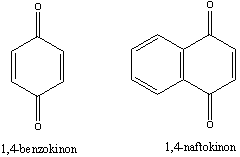
A kinonok oxocsoportjai reverzibilisen redukálhatók dihidroxi-származékká:
A kinonok elterjedt vegyületek a növényvilágban és az állati szervezetekben is. A benzokinon típsú kinonok közül az ubikinon vagy más néven koenzim-Q az elektrontranszport lánc lényeges eleme, a mitokondrium apoláris részéhez kapcsolódik a 6-10 tagú izoprénegységnek köszönhetően:
Az 1,4-naftokinon izoprénoldalláncot tartalmazó származékai a K-vitaminok. A K-vitaminok hiánya melegvérű állatokban a véralvadás zavarát okozza:
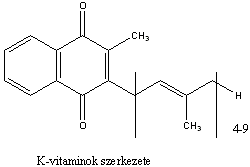
1., Nevezze el az alábbi vegyületeket az IUPAC szabályai szerint
2., Írjon példát a következőkre:
a., acetál
b., geminális diol
c., imin
d., félacetál
3., Milyen termékek keletkeznek, ha a fenil-acetaldehidet (C6H5CH2CHO) az alábbi reagensekkel reagáltatjuk?
a., Tollens-reagens
b., CH3OH, H+-katalizátor
c., NH2OH
4., Rajzolja fel az alábbi molekulák deprotonálódásával keletkező enolátiont!
5., Melyik vegyület esetében játszódik le aldol-kondenzáció? Írja fel a keletkezett terméket!
a., ciklohexanon
b., 2,2,6,6-tetrametil-ciklohexanon
c., benzaldehid
d., formaldehid
6., Írja fel az alábbi vegyületek aldol-termékeit!
7., A fahéjaldehidet a benzaldehid és az acetaldehid vegyes aldolreakciójával lehet előállítani! Írja fel a reakciót! Milyen más termék keletkezhet a reakcióban?
Tartalom
Tartalom
A pohidroxi-aldehideket aldózoknak, a polihidroxi-ketonokat ketózoknak nevezik. A szénatomok száma szerint beszélhetünk triózokról, tetrózokról, pentózokról, hexózokról. A monoszacharidok kristályos, vízben jól oldódó, édes ízű vegyületek, a dihidroxi-aceton kivételével optikailag aktívak.
A félacetál képződés során 5- vagy 6-tagú, oxigénatomot tartalmazó heterociklusok keletkeznek. Az 5-tagú gyűrűs szénhidrátokat a furán után furanózoknak, a 6-tagú gyűrűs szénhidrátokat a 2H-pirán alapján piranózoknak nevezik.
A monoszacharidokból az átkristályosítás módjától függően kétféle, optikailag aktív módosulat nyerhető. Így a D-glükóz vizes oldatából ecetsav jelenlétében olyan kristályokat kapunk, amelynek frissen készített vizes oldata [α]=+112º fajlagos forgatóképességgel rendelkezik. Ez az érték folyamatosan csökken, +52,7º-ra. A D-glükóz vizes oldatát piridin jelenlétében kristályosítva +18,7º fajlagos forgatóképességű módosulatot kapunk. Frissen készített vizes oldatának forgatóképessége folyamatosan nő, és végül ez is +52,7º-nál állapodik meg.
A kétféle D-glükóz módosulat optikai aktivitásában leírt különbséget és változásukat egy egyensúlyi állapotig mutarotációnak nevezzük. Ennek magyarázatát Tollens adta meg: a cukrokban a karbonilcsoport és a hozzá kedvező térhelyzetű alkoholos hidroxilcsoport között intramolekuláris nukleofil addíció játszódik le, félacetál képződik (ld. oxovegyületek reakciói).
A gyűrűs félacetál szerkezet kialakulásakor az eredetileg sp2 hibridállapotú karbonil szénatom aszimmetriacentrummá válik, egy új, ún. glikozidos hidroxilcsoportot tartalmaz. Ennek megfelelően a gyűrűs forma keletkezésekor két sztereoizomer alakulhat ki. Ezeket a vegyületpárokat anomereknek nevezik, és α vagy β jellel különböztetik meg. Az α-anomerben a glikozidos hidroxilcsoport és a hidroximetil csoport ellentétes térfélen, a β-anomerben azonos térfélen találhatók.
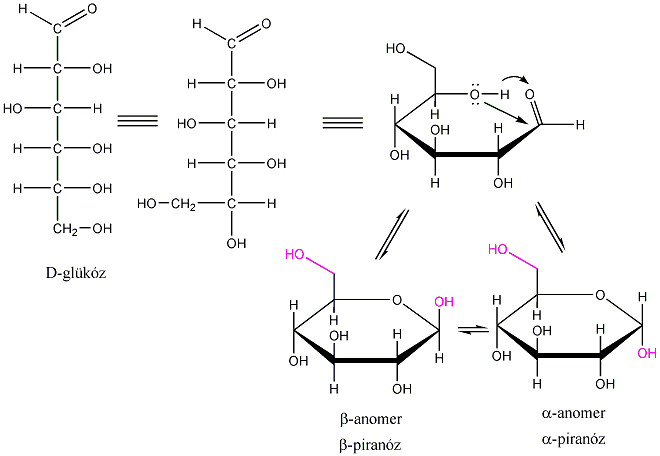
Az anomerek diasztereomer vegyületpárok, tehát forgatóképességük, fizikai és kémiai tulajdonságaik is eltérnek egymástól. Az anomerek és a nyíltláncú alak vizes oldatban egyensúlyi elegyet alkotnak, egymásba átalakulnak.
A D-glükóz vizes oldatának egyensúlyi elegye mintegy 36 % α-anomert, 63 % β-anomert és csak 1 %-nyi nyíltláncú formát tartalmaz. Ez az arány azzal magyarázható, hogy az α-anomerben a glikozidos hidroxilcsoport axiális térállású, és így két axiális hidrogénatommal térben közel kerülnek egymáshoz. A β-anomerben a nagy tértöltésű hidroxilcsoportok és hidroximetil csoport mindegyike ekvatoriális helyzetű, ami energetikailag kedvezőbb.
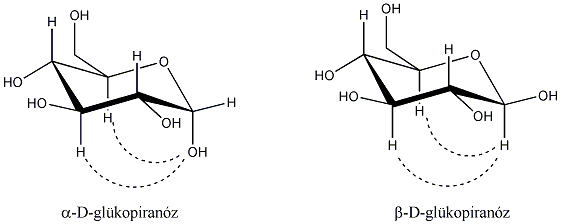
Mind az aldózok, mind a ketózok enyhe redukálószerek hatására alkoholokká alakulnak. A redukció csak a nyíltláncú, karbonilcsoportot tartalmazó formában játszódik le, a keletkező cukoralkoholok is nyíltláncúak lesznek. A D-glükózból így D-szorbitol keletkezik:
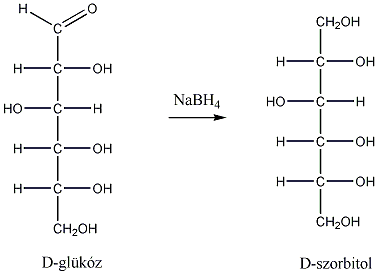
Oxidációs reakcióban az aldehidcsoport oxidációjával az aldózokból aldonsavak jönnek létre. További erélyes oxidációra a primer alkoholos hidroxilcsoport is átalakul karboxilcsoporttá, aldársavak keletkeznek.
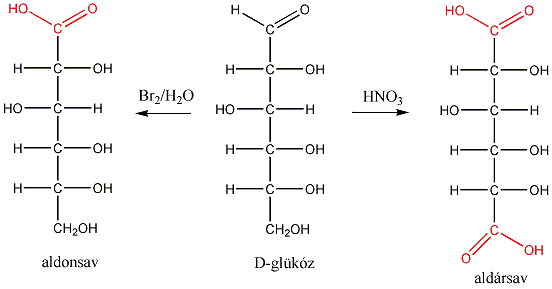
Az uronsavakban a primer alkoholos hidroxilcsoport oxidálódik, ezért a gyűrűs félacetál szerkezet megmarad. Az uronsavak többféle bioaktív molekula felépítésében vesznek részt, így például poliszacharidok (pektin) alkotórészei.
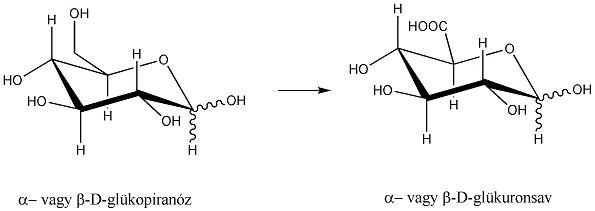
A monoszacharidok kimutatása legegyszerűbben lúgos közegben végzett oxidációval – Fehling- és Tollens-próbával – végezhető el. A karbonilvegyületek reakciójánál felírt reakcióegyenletekhez hasonlóan itt is az aldehidcsoport karboxilcsoporttá oxidálódik, míg a fémion redukálódik. A karbonilvegyületek közül a ketonok nem adják ezeket a reakciókat, ellenben a ketózok az alább ismertetett epimerizácóval egyensúlyi reakcióban aldózokká alakulnak, így pl. a fruktóz is adja a Fehling-próbát.
Azokat a monoszacharidokat, amelyek konfigurációja csak egy szénatomon különbözik, epimereknek nevezzük. Az epimerizáció biológiai folyamatokban is lejátszódik, ennek során a glükóz, a fruktóz és a mannóz enol-oxo tautomériával egymásba egy közös endiol formán keresztül átalakulnak:
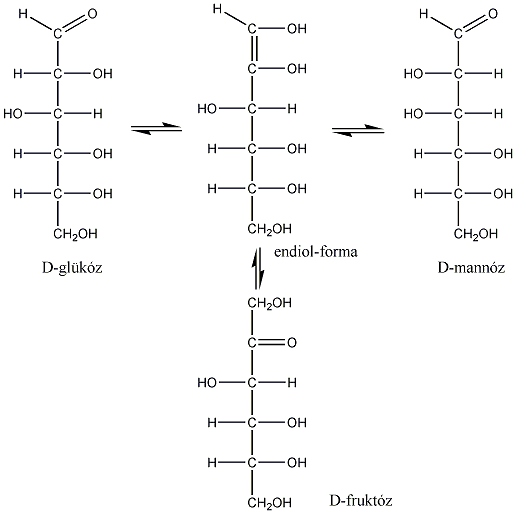
A monoszacharidok foszfátészterei minden sejtben megtalálhatók, a szénhidrát anyagcsere intermedierjei. A fotoszintézisben a CO2 megkötésében játszanak szerepet. Így a ribulóz-1,5-difoszfát a CO2 megkötésével egy hatszénatomos cukorfoszfáttá alakul, majd ebből több lépésen keresztül a fruktóz-1,6-difoszfát szintéziséhez szükséges D-glicerinaldehid-foszfát keletkezik. Jelentős szerepük van a szénhidrátok biológiai lebomlása során bekövetkező energia felszabadításában és tárolásában.
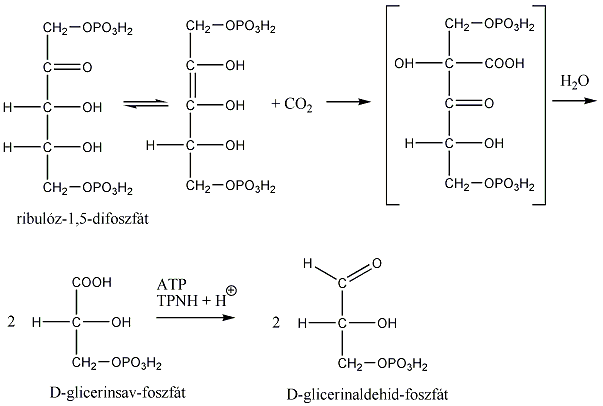
A poliszacharidok felépítésében szulfátészterek is előfordulnak, pl. a véralvadást gátló heparinban vagy a kötőszövetekben lévő kondroitinban.
Az O-glikozidok keletkezése során a monoszacharid glikozidos hidroxilcsoportja vízkilépés közben kapcsolódik egy másik hidroxilcsoportot tartalmazó molekulával, így acetál képződik.
Az N-glikozidokban a glikozidos hidroxilcsoport egy másik molekula amino- vagy iminocsoporttal alkot vízkilépéssel C-N kötést (ld. nukleozidok). A cukorrészhez kapcsolódó másik molekulát aglikonnak nevezik.
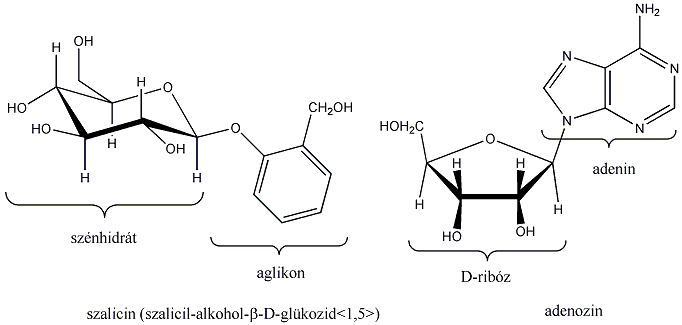
A triózok közé tartoznak a D-glicerinaldehid és a dihidroxi-aceton. Mindkét vegyület fontos szerepet játszik a szénhidrátok lebontásában és szintézisében (ld. karbonilvegyületek).
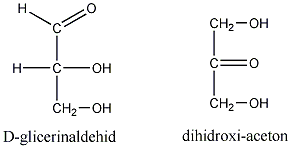
A D-ribóz a ribonukleotidok és ribonukleinsavak építőköve, ezért minden növényi és állati sejtben megtalálhatók. A furanózgyűrűs alak glikozidos hidroxilcsoportja a purin ill. pirimidinbázis nitrogénatomjához kapcsolódik, N-glikozidot létrehozva. Vitaminokban és koenzimekben is megtalálható.
A 2-dezoxi-D-ribóz a dezoxiribonukleinsavak alkotórésze. Elsősorban a sejtmagban található, N-glikozidos kötéssel kapcsolódik a nukleinbázisokhoz.
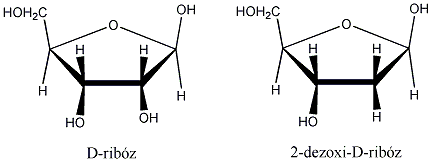
A D-glükóz a legfontosabb szénhidrátok egyike. Szabad állapotban gyümölcsökben, kötött állapotban diszacharidokban és poliszacharidokban is gyakori.
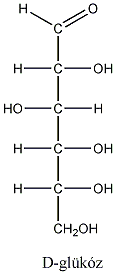
A D-mannóz a növényekben gyakori mannánok építőköve.
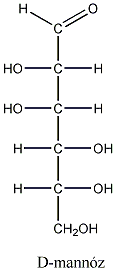
A D-galaktóz kötött állapotban a növény- és állatvilágban is igen gyakori. Így megtalálható a laktózban és az idegsejtekben előforduló szfingolipideknek is alkotórésze.
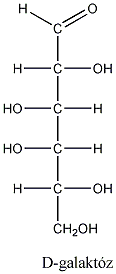
A D-fruktóz a növényvilágban igen gyakori szabad állapotban is, például gyümölcsökben vagy mézben. Kötött állapotban a szacharóz alkotórésze.
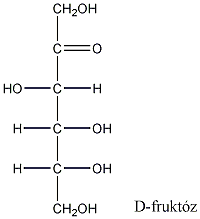
Az L-aszkorbinsav, a C-vitamin is cukorszármazék. Két enolos hidroxilcsoport található benne, ezért a gyűrűs, lakton formája is savas jellegű. A C-vitamin redukáló hatású vegyület, nagyon könnyen oxidálódik, L-dehidroaszkorbinsavvá alakul. A reakció reverzibilis, ezért a sejtek redoxireakcióiban vesz részt.
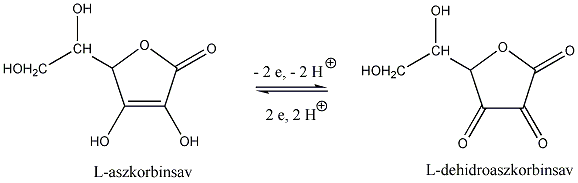
Az emberi szervezet nem tudja szintetizálni, a táplálékkal, elsősorban zöldségekkel és gyümölcsökkel jut be a szervezetbe. Hiányában skorbut alakul ki.
Tartalom
A diszacharidok glikozid típusú vegyületek, két monoszacharidból vízkilépéssel kapcsolódnak össze. Vízben jól oldódnak, édes ízűek. Fehling-reakcióban mutatott viselkedésük alapján osztályozzák őket.
A nem redukáló diszacharidok nem tartalmaznak szabad glikozidos hidroxilcsoportot. Összekapcsolódásukkor a két monoszacharid glikozidos hidroxilcsoportja lép kondenzációs reakcióba. Ezek a diszacharidok nem adják a redukciós cukorkimutatási próbákat és oldatuk mutarotációt sem mutat.
A redukáló diszacharidok keletkezése során az egyik molekula glikozidos hidroxilcsoportja reagál a másik molekula valamelyik alkoholos hidroxilcsoportjával. Mivel ezekben a diszacharidokban van szabad glikozidos hidroxilcsoport, ezért redukciós reakciókban reagálnak és oldatuk mutarotál is.
A szacharóz gyakorlati szempontból a legjelentősebb nem redukáló diszacharid. Nagy mennyiségen állítják elő és alkalmazzák élelmezési célokra. A növényvilágban elterjedt, ipari méretű kivonásához mérsékelt éghajlatú országokban a cukorrépa, trópusi országokban pedig a cukornád jön számításba. A szacharózban egy α-D-glükopiranóz és egy β-D-fruktofuranóz kapcsolódik össze a glikozidos hidroxilcsoportokon keresztül, 1-2 kötést alakítva ki.
A szacharóz nem csak enzimek hatására, hanem savas közegben is monoszacharidjaira hidrolizál. Ezt a folyamatot redukciós reakción vagy a forgatóképesség változásán keresztül is követhetjük. A forgatóképesség a hidrolízis során folyamatosan csökken, eléri a 0 értéket, majd az oldat balra forgatóvá válik. A forgatóképesség előjelének megváltozása miatt a szacharóz hidrolízisét invertálásnak nevezzük, a képződő glükóz és fruktóz elegyét invert cukornak. A jelenség arra vezethető vissza, hogy a mutarotáció megállapodása után a balra forgató D-fruktóz ([α] = -92,4º) fajlagos forgatóképessége jóval nagyobb, mint a jobbra forgató D-glükózé ([α] = +52,7º).
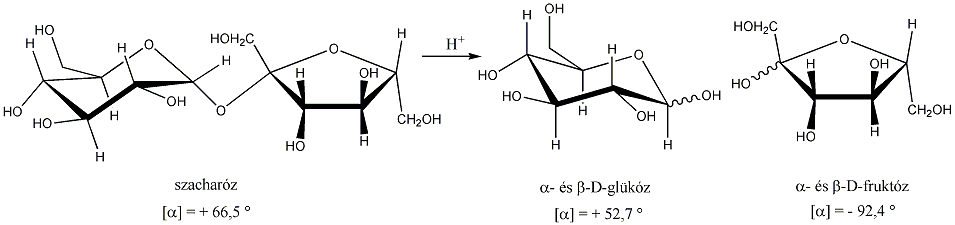
A redukáló diszacharidok közül a maltóz molekulájában két D-glükóz kapcsolódik össze α-glikozidos 1-4 kötésben. A keményítő hidrolízisterméke, a keményítő enzimatikus lebontása során keletkezik.
A cellobióz szabadon nem fordul elő. A legelterjedtebb szénvegyületnek, a cellulóznak az alkotórésze, ebből állítható elő részleges hidrolízissel. Két D-glükózból β1-4) kötéssel épül fel. Szerkezetileg rokon a maltózzal, de a glikozidos hidroxilcsoportok konfigurációjában való különbség a molekula térszerkezetében jelentős eltérést okoz:
A laktóz D-glükózból és D-galaktózból felépülő diszacharid, a monoszacharidok 1-4 kötéssel kapcsolódnak egymáshoz. Emlősök tejében 5-8 % mennyiségben fordul elő.
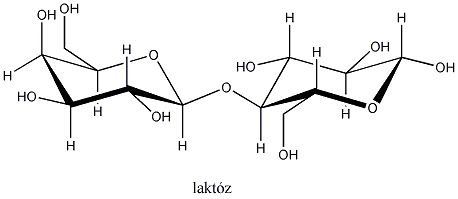
A poliszacharidok nagy molekulatömegű, monoszacharid egységekből felépülő vegyületek. Vízben nem oldódnak, vagy kolloid oldatot képeznek. Funkciójuk alapján két csoportba sorolják őket: tartalék tápanyagok és vázanyagok.
A keményítő a növényvilágban elterjedt tartaléktápanyag. A fotoszintézis helyén keletkezik, majd a növényi nedvekkel kerül a gumókba, gyökerekbe és magokba. A raktározott keményítőt a növény újra fel tudja használni a polimer molekula lebontásával. A keményítő két eltérő szerkezetű anyagból, amilózból és amilopektinből áll. Az amilózban a glükózegységek α(1-4) kötéssel kapcsolódnak össze, a kialakult lánc elágazásokat nem tartalmaz. Az amilózlánc helikális, egy csavarmenetre hat glükózegység jut. A hélixszerkezetet intramolekuláris hidrogénkötések stabilizálják. Az amilóz jóddal intenzív kék színeződést ad, ami azon alapszik, hogy a jódmolekulák és a hélix zárványvegyületet képeznek, megváltoztatva a jódmolekula eredeti fényabszorpcióját.
Az amilopektinben is az α(1-4) kötések dominálnak, de 12-20 egységenként α(1-6) elágazások is vannak.
A glikogén az állati szövetek tartalék tápanyaga. Jelentős mennyiségben raktározódik a májban és az izmokban, szerkezete az amilopektinére hasonlít.
A cellulóz a bioszféra szerves anyagának közel a felét teszi ki, a növényi rostok fő alkotórésze. D-glükóz egységekből β(1-4) kötésekkel épül fel. Vízben nem oldódik, 3000-10000 glükózegységből áll. Enyhe savas hirolízissel cellobiózra bonthó. A molekula hosszú láncokat alkot, amelyet intramolekuláris hidrogénkötések stabilizálnak.
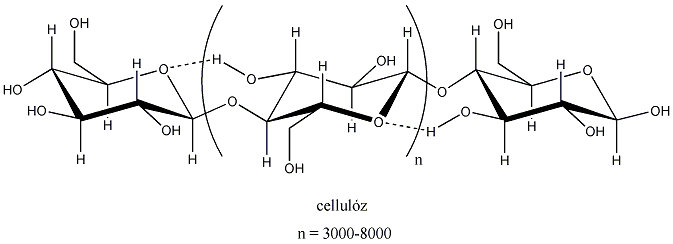
A cellulózmolekulák párhuzamosan futó kötegekbe is rendeződnek, az egyes fonalak között intermolekuláris hidrogénkötések alakulnak ki. Ez a nagyfokú rendezettség mechanikai szilárdságot kölcsönöz a cellulóznak. A cellulóz biológiai lebomlását a celluláz enzim katalizálja.
A pektinek elsősorban a növények lágyabb szöveteiben fordulnak elő. Felépítésükben D-galaktóz és D-arabinóz vesz részt. Forró vízben gélt képez, lehűtve zselét vagy kocsonyát alkot, ezért az élelmiszeriparban használják.
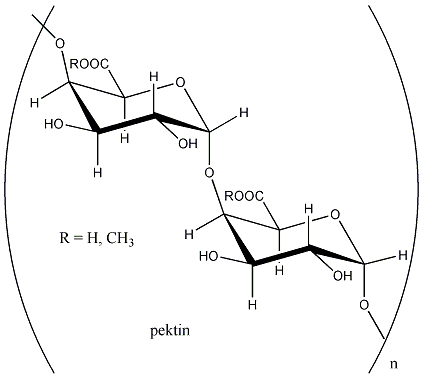
A kitin az ízeltlábúak külső vázát és a gombák sejtfalát alkotja, N-acetil-glükózamin egységekből épül fel:
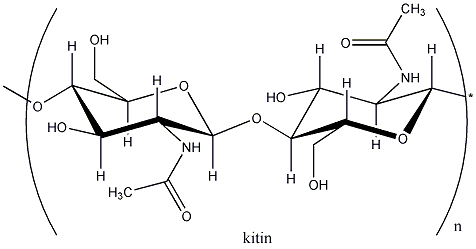
1., Definiálja a következő fogalmakat és írjon mindegyikre példát!
a., monoszacharid
b., glikozidos hidroxilcsoport
c., redukáló cukor
d., D-cukor
e., piranóz-gyűrűs forma
f., 1,4’-kapcsolódás
2., Rajzolja fel a D-ribulóz 5-tagú-gyűrűs félacetál formáját!
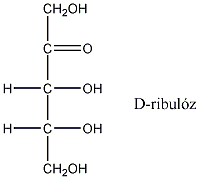
3., Milyen sztereokémiai összefüggés van a D-glükóz és a D-galaktóz között? Milyen megállapításokat lehet tenni az alábbi fizikai sajátságaikra?
a., olvadáspont
b., vízoldhatóság
c., sűrűség
d., fajlagos forgatóképesség
4., Rajzolja fel a lehetséges D-2-ketohexózok szerkezetét!
5., A glükóz mellett más monoszacharidok is mutatják a mutarotáció jelenségét. Az α-D-galaktopiranóz fajlagos forgatóképessége [α]D=+150.7°, a β-D-galaktopiranóz fajlagos forgatóképessége [α]D = + 52,8 °, az egyensúlyi elegyé [α]D = + 80.2 °. Mekkora az egyes anomerek aránya az egyensúlyi elegyben?
Tartalom
- 23. Karbonsavak
- 24. A karbonsavak és karbonsavszármazékok elektronszerkezete:
- 25. Karbonsavak csoportosítása:
- 26. A karbonsavak és karbonsavszármazékok elnevezése:
- 27. Karbonsavak és karbonsavszármazékok előállítása
- 28. A karbonsavak, karbonsavszármazékok fizikai tulajdonságai
- 29. A karbonsavak és karbonsavszármazékok kémiai tulajdonságai
- 30. A karbonsavak és karbonsavszármazékok fontosabb képviselői:
- 31. Szénsavszármazékok
Karbonsavaknak a karboxilcsoportot tartalmazó vegyületet nevezzük. A karbonsav hidroxilcsoportjának és /vagy karbonilcsoprtjának más csoportra történő cseréjével karbonsavszármazékokhoz jutunk. Legfontosabb karbonsavszármazékok:
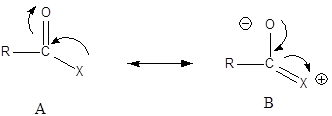
A karbonilcsoport szénatomja sp2 hibridállapotú, a hozzákapcsolódó hidroxilcsoporttal vagy heteroatommal, az R csoporttal és a karbonil oxigénnel létesít egy-egy σ-kötést, a fennmaradó pz pályán levő elektron pedig a karbonil oxigénnel létesít egy π-kötést. A karbonilcsoport (C=O) kettőskötése és a hidroxilcsoport oxigénje vagy a heteroatom nemkötő elektronpárja konjugációs kölcsönhatást létesít, vagyis egy tricentrikus, négyelektronos rendszer jön létre amely A és B határszekezettel jellemezhető:
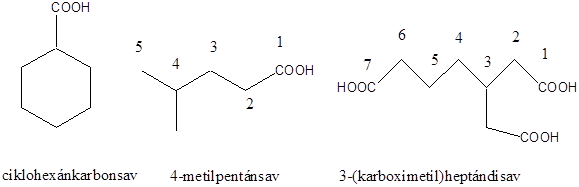
Az A határszerkezet inkább a karbonsavakra és karbonsavhalogenidekre jellemző, míg a B határszerkezet hozzájárulása inkább az észterek és a savamidok esetében a nagyobb.
Beszélhetünk mono-, di-, tri- és polikarbonsavakról, ezen belül 1,2- vagy 1,3- vagy 1,4-dikarbonsavakról. A karboxilcsoporthoz kapcsolódó szénlánc jellege szerint lehetnek alifás telített, telítetlen, gyűrűs, aromás ill. heteroaromás karbonsav.
Tartalom
Ha a karboxilcsoport előtagként szerepel, akkor a karboxi, míg utótagként - ha a karboxilcsoport szénatomja része az alapvegyületnek, akkor a sav, ha nem akkor a karbonsav elnevezést használjuk. A karboxilcsoport az elnevezésnél elsőbbséget élvez a karbonsavszármazékokkal (anhidrid, észter, savhalogenid, amid) szemben.
Közvetlenül a gyűrűhöz kapcsolódó karboxilcsoport, vagy kettőnél több karboxilcsoportot tartalmazó nyílt láncú vegyület esetében az elnevezés az alapvegyületként megadott szénhidrogén nevéhez fűzött karbonsav utótaggal történik:
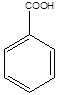
Sok egyszerű karbonsavat latin vagy görög eredetű, ill. triviális névvel illetnek. A leggyakoribb triviális nevet, ill. a belőle származtatható acilcsoport nevét az alábbi táblázatban ismertetjük:
26.1. táblázat -
|
Képlet |
Sav neve |
Acilcsoport neve |
|
HCOOH |
hangyasav |
formil |
|
CH3COOH |
ecetsav |
acetil |
|
CH3CH2COOH |
propionsav |
propionil |
|
CH3(CH2)2COOH |
vajsav |
butiril |
|
CH3(CH2)3COOH |
valeriánsav |
valeril |
|
CH3(CH2)4COOH |
kapronsav |
kaproil |
|
CH3(CH2)14COOH |
palmitinsav |
palmitoil |
|
HOOC-COOH |
oxálsav |
oxalil |
|
HOOC-CH2-COOH |
malonsav |
malonil |
|
HOOC-(CH2)2-COOH |
borostyánkősav |
szukcinil |
|
HOOC-(CH2)3-COOH |
glutársav |
glutaril |
|
H2C=CHCOOH |
akrilsav |
akriloil |
|
|
benzoesav |
benzoil |
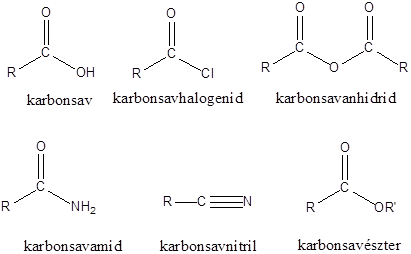
A karbonsavakból kétféle csoport vetethető le, az acilcsoport (ld. fenti táblázat) és a karboxilcsoport hidrogénjének eltávolításával kapott savmaradék, amely savra végződő név esetén oát, karbonsavra végződő név esetén karboxilát végződéssel nevezünk el. A savmaradékkal képezzük a karbonsavak sóinak a nevét oly módon, hogy a kation kerül előre, majd azt követi a savmaradék:
A csoportfunkciós nómenklatúra szabályai szerint történik: acilcsoport neve + halogenid. Előtagként a halogénkarbonil elnevezést használjuk:
A karbonsavanhidridek a karbonsavakból vezethetők le, mégpedig a karboxilcsoport hidrogénjének az aciloxi- vagy aroiloxicsoportra történő cseréjével. Az azonos karbonsavakból nyert anhidrideket egyszerű anhidrideknek nevezzük, a különböző karbaonsavakból nyert anhidridek a vegyes anhidridek. Elnevezés a karbonsav nevének sav utótagja savanhidridre módosul.
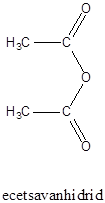
Az észterek elnevezése a csoportfunkciós névalkotásssal történik. Az észterek csoportfunkciós nevében amennyiben az észtercsoport a főcsoport az alkohol/fenol alkil/aril csoportjait tüntetjük fel utána pedig a karbonsavkomponens savmaradékát. Az ún. körülírásos nevet kerüljük.
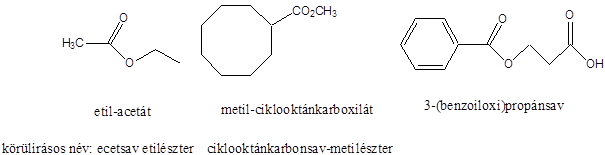
Ha az észtercsoportnál magasabb rendű funkcióscsoport is van a molekulában akkor az utóbbi lesz a főcsoport akkor az észtercsoportot alkil/ariloxikarbonil előtaggal kell megadni.
A savamidok elnevezése: amid ill. karboxamid ha utótag, előtagként karbamoil vagy acilamino elnevezést használjuk. Az amid nitrogénen szubsztituált amidokat N-szubsztituált amidként nevezzük el. Ha az amin anilin, akkor anilid utótagot használjuk.
Az azonos acilcsoporttal helyettesített amidokat diacilaminként (kivéve: di- és triacetamid), gyűrűs diacilaminokat imidként nevezzük el.
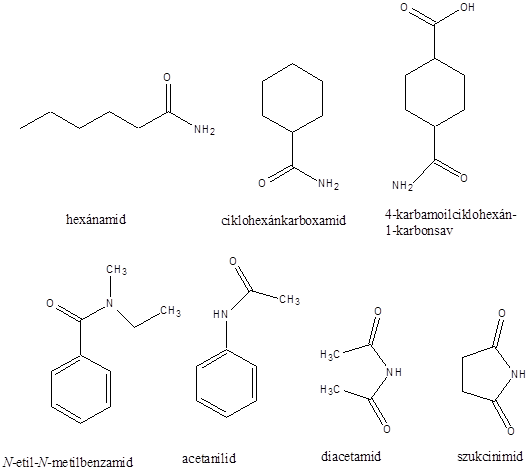
Tartalom
Karbonsavakat elő lehet állítani primer alkoholok oxidációjával kálium-permanganát, króm-trioxid, hidrogén-peroxid, levegő oxigénje Co(III)-só jelenlétében. Értelemszerűen köztitermékként aldehid keletkezik, de ha aldehidből indulunk ki, akkor enyhébb oxidálószer pl. AgNO3/NH4OH is elegendő a karbonsavvá történő oxidáláshoz. Alkilbenzolok savas kálium-permanganáttal szintén karbonsavat adnak.
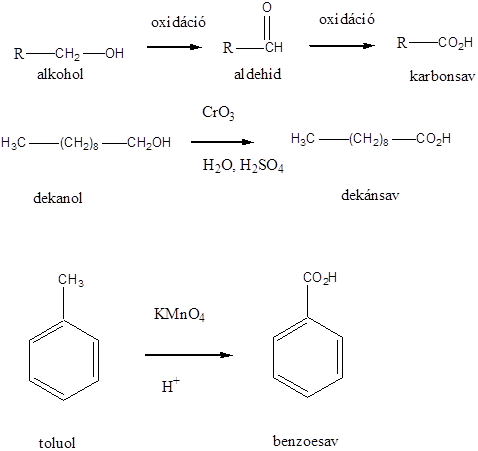
A karbonsavak előállíthatók még anionos jellegű szénatomot tartalmazó fémorganikus vegyület (pl. Grignard-reagens) és szén-dioxid reakciójával, ilyenkor 1-gyel nagyobb szénatomszámú karbonsavak képződnek. A nitrilek és az észterek hidrolízisével szintén karbonsavak nyerhetők:
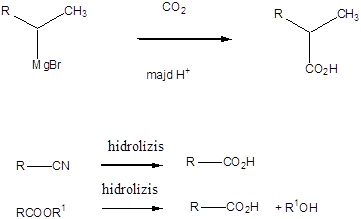
A savkloridokat karbonsav és szulfinil-diklorid reakciójával állítják elő:
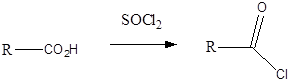
Karbonsavak ecetsavanhidriddel vagy foszfor-pentoxiddal történő vízelvonással szintetizálhatók. Vegyes anhidridek a savkloridból és a másik savkomponens sójából nyerhetjük:
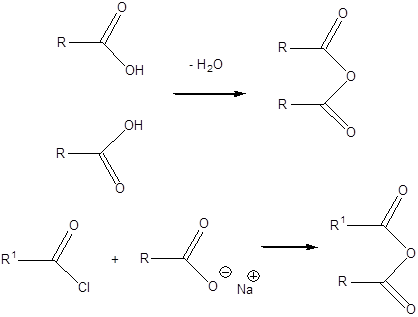
Karbonsav és alkoholok és ásványi savak jelenlétében végzett reakcióját Fischer-féle észteresítésnek nevezzük. Az egyensúlyi folyamat azonban a víz eltávolításával, ill. a kiindulási anyagok koncentrációjának növelésével a képződés irányába eltolható.
Savkloridok és alkohol reakciójával, valamint karbonsavak sóinak alkilezésével szintén észterekhez juthatunk
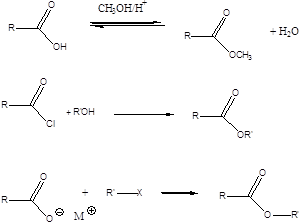
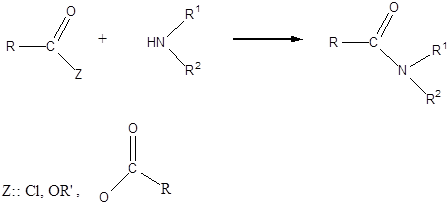
A karbonsavak egyes vegyületcsoportjai homológ sort alkotnak, így forráspontjuk a szénatomszám növekedésével növekszik. Fizikai tulajdonságaikat a szénatomszám mellett a funcióscsoportok száma, a kialakuló hidrogénkötések száma, valamint a szénelágazások befolyásolják. Az egyenes szénláncú karbonsavak első kilenc tagja szobahőmérsékleten folyadék a molekulák fonalszerű asszociációjának köszönhetően. A gőzfázisban a karbonsavak dimereket képeznek.
A karbonsavak olvadáspontja a szénatomszámmal nem az lineárisan növekszik, az olvadáspont szabályszerű emelkedése csak akkor állapítható meg, ha páratlan, ill. páros szénatomszámú karbonsavakat külön vizsgáljuk. A páros, ill. páratlan számú karbonsavak térkémiai okok miatt más-más kristályrendszerben kristályosodnak.
Monokarbonsavak olvadáspontjának változása a szénatomszám függvényében
A hangyasav, ecetsav, propionsav, vajsav vízzel korlátlanul elegyedik, a szénatomszám növelésével a hidrofób tulajdonság erősödik, ezért ezek a karbonsavak vízben nem, csak lipofil oldószerkben oldódnak.
A dikarbonsavak szilárd halmazállapotú vegyületek, vízben jól oldódnak. A karbonsavhalogenidek alacsony forráspontú anyagok vízzel heves reakcióba lépnek. A karbonsavanhidridek forráspontja magasabb a megfelelő savhalogenid forráspontjánál.
Az észterek forráspontja alacsonyabb, mint a megfelelő karbonsav forráspontja, vízzel nem elegyednek, az alacsony szénatomszámú karbonsavak észterei kellemes gyümölcs illatúak. A karbonsavamidok a formamid kivételével jól kristályosodó vegyületek, asszociátumokat képeznek hidrogénkötéseik révén, ezért forráspontjuk magas.
Tartalom
A karbonsavak reaktivitása három fő reakció szerint csoportosítható:
- Sav-bázis reakciók
- A karbonilcsoport nukleofil reagensekkel végbemenő reakciói
-A karbonilcsoport α-szénatomján végbemenő reakciók
A karbonsavak erősebb savak mint az alkoholok, de gyengébb savak mint az ásványi savak:
Néhány sav erőssége
29.1. táblázat -
|
Név |
pKa |
|
HCl (sósav) |
-7 |
|
CCl3CO2H |
0,64 |
|
HCO2H |
3,75 |
|
C6H5CO2H |
4,19 |
|
H2C=CHCO2H |
4.25 |
|
CH3CO2H |
4.75 |
|
CH3CH2OH |
16 |
A savak erősségét több tényező is befolyásolja. A helyettesítetlen monokarbonsavak közül a legerősebb sav a hangyasav. Ha a hidrogént elektronküldő metilcsoportra cseréljük, kb. egy nagyságrenddel csökken a savi erősség. Ez nem csak a metilcsoport elektronküldő jellegéből fakad, hanem abból is, az acetátiont a víz kevésbé jól szolvatálja mint a formiát iont. Elektronvonzó szubsztituens bevezetése az α-helyzetű szénatomra növeli az aciditást, pl. a triklórecetsav erősebb sav, mint az ecetsav, mert a klóratomok nagyobb elektronegativitása polározottabbá teszi az O-H kötést. Távolabbi szubsztitúció az aciditást alig befolyásolja.
A dikarbonsavak disszociációja két lépében játszódik le, ezért ezeket pKa1 és pKa2 értékkel jellemezzük. A pKa1 kisebb érték (az első proton könnyebben disszociál) a pKa1 és pKa2 közti különbség annál nagyobb, minél közelebb van a két karboxilcsoport egymáshoz a molekulában. A kisebb pKa2 értéket azzal magyarázhatjuk, hogy egy negatív töltésű, részben disszociált molekulától már elektrosztatikusan kedvezőtlen lehasítani egy pozitív töltésű részecskét.
Az oxovegyületekhez hasonlóan a karboxilcsoport szén-oxigén kettőskötése is erősen polarizált. A polarizáltság és a polarizálhatóság eredményeként a karbonilcsoport szénatomján nukleofil reagensekkel támadható, majd a képződött tetraéderes intermedier eliminációs reakcióban stabilizálódik, vagyis a karbonsavak és származékaik nukleofilekkel addíciós-eliminációs mechanizmusú szubsztitúciós reakció szerint reagálnak.
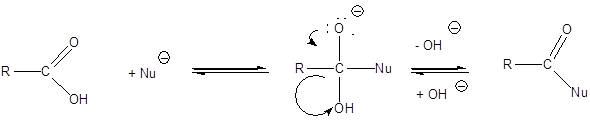
A kétlépcsős reakciónak a nukleofil addíció a meghatározó lépése és azért a reakciót nukleofil acilszubsztitúciónak nevezzük (S N Ac). Az egyes savszármazékok reaktivitását két, ellentétes hatás irányítja: az X-szubsztituens –I effektusa –ez minél nagyobb annál nagyobb a karbonilszénatom elektrofilitása és nő a származék acilező készsége. Az X heteroatom +M effektusa, vagyis a nemkötő pár és a C=O kötés π-elektronjainak delokalizációja minél nagyobb, annál kisebb a reakciókészség
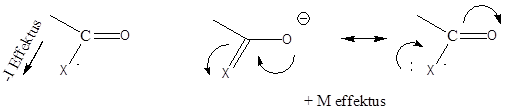
Ebből adódik a savszármazékok reakciókészsége:
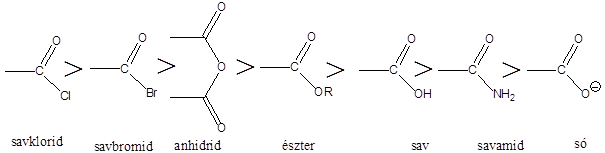
A karbonsavak alkoholokkal savkatalizált reakcióban észtereket képeznek. A reakciót a karbonsav gyors protonálódása vezeti be, amit az alkohol nukleofil addíciója követ, majd az addukt vízkihasadásaal stabilizálódik:
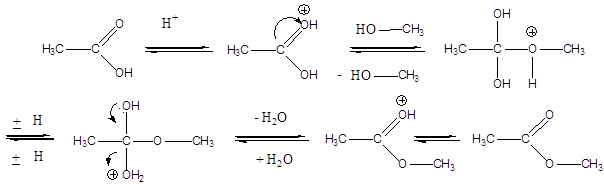
Az ellenkező irányú folyamat az észterek savas hidrolízise. Mivel a reakció savkatalizált és acilhasadással játszódik le, továbbá a sebességmeghatározó lépése bimolekuláris azért ezt a reakciót A Ac 2 megjelöléssel illetjük.
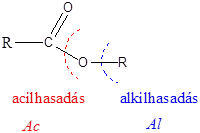
A BAc2 átalakulásnál sztöchiometrikus mennyiségű bázis szükséges, amikor is a hidroxidion beépül a tetraéderes intermedierbe, majd az X- csoport eliminálódik. Tulajdonképpen ez a folyamat az észterhidrolízis:
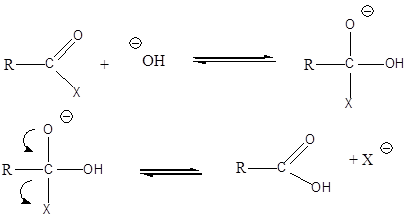
A monomolekuláris AAl1 reakció bevezető lépése a karbonilcsoport protonálódása, majd lassú folyamatban az alkoholrész alkil csoportja hasad le. A képződött karbokationból alkén vagy alkohol lesz.
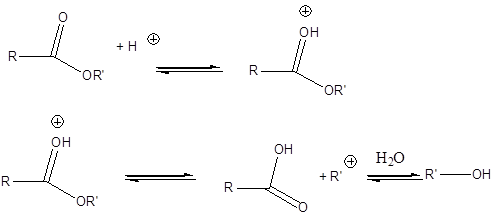
Az α-hidrogént tartalmazó karbonsavszármazékok CH-savaknak is tekinthetők. Általában CH-savaknak nevezzük azokat a vegyületeket, amely egy vagy két elektronszívó csoportot tartalmaznak, amelyekről így proton válhat le A CH-savak protonjának lehasításához erős bázis szükséges. A karbonsavak ilyen irányú reaktivitása kicsi, ugyanis első lépésben karboxilát ion keletkezik, amely csökkenti az α-CH savasságát, a savamidok reaktivitása kicsi, a karbonsavhalogenidek nagy reakció készségük miatt erős bázisokban más típusú reakciót adnak. Preparatív szempontból a karbonsavnitrilek és a karbonsavészterek α-CH proton lehasításának van jelentősége. Ilyen reakció a Claisen-kondenzáció, amelyben két észtemolekula vesz részt. Az észterből képzett karbanion a másik észtermolekula karbonilcsoportjával reagál. A molekulából etanol kihasadása után, majd az enol tautomerizációját követően egy β-oxoészter alakul ki.
Karbonsavakból elemi halogénnel, sztöchiometrikus foszfor jelenlétében (Hell-Volhard-Zelenszkij-reakció) α-halogénsav-halogenidek nyerhetők, melyek a feldolgozáshoz használt vízzel α-halogénkarbonsavvá alakulnak. Feleslegben alkalmazott halogén jelenlétében α,α-dihalogén ill. α,α,α-trihalogén karbonsavvá alakulnak.
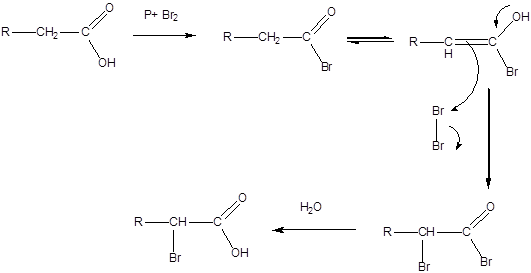
Az α-halogénkarbonsavak a halogénatom elektronszívó hatása miatt erősebb savak mint a nem halogénezett analogonjaik. A halogénezett karbonsavak nukleofilekkel nukleofil szubsztitúciós és /vagy eliminációs reakcióba lépnek. Így pl. α-halogénkarbonsavak vizes nátrium-hidroxiddal szubsztitúciós reakcióban α-hidroxikarbonsavat képeznek, ugyanakkor α,α-dihalogénkarbonsavak lúgos hidrolízise α-oxokarbonsavat ererdményez.
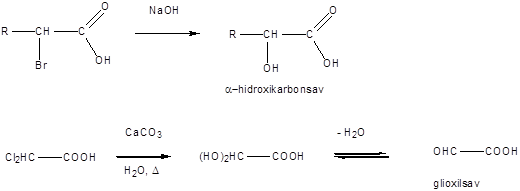
Az oxovegyületkből könnyen nyerhető ciánhidrinek hidrolízise szintén α-hidroxikarbonsavat eredményez, ez analóg a Stecker-szintézissel, amely α-aminosavhoz vezet:
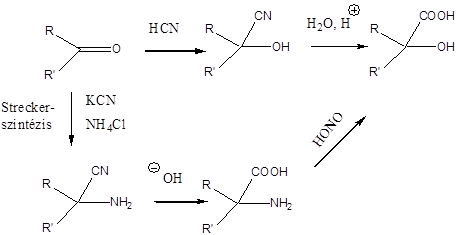
Az α-hidroxisavak erősebb savak mint a megfelelő karbonsavak a hidroxilcsoport elektronszívó sajátsága miatt. Az α-hidroxikarbonsavak melegítés hatására hattagú gyűrűs észterekké, dilaktonokká alakulnak. Az α-hidroxikarbonsavak híg ásványi savval melegítve széndioxidot veszítenek és ezáltal egy szénatommal rövidebb aldehid keletkezik.
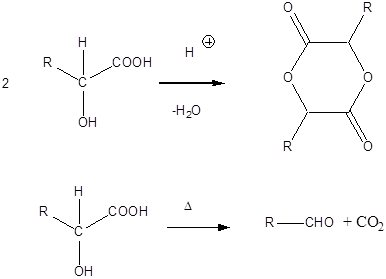
Az oxosavakra mind a karbonsavak, mind az oxovegyületek reakciói jellemzők, de reaktivitásuk a karboxilcsoport és az oxocsoport egymáshoz viszonyított helyzetétől függ. Az α-oxovegyületek már enyhe oxidáló szer hatására is széndioxiddá és eggyel rövidebb szénatomot tartalmazó karbonsavvá alakulnak, pl:
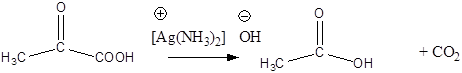
Szintetikus szempontból a Claisen-kondenzációval elérhető β-oxosavak és észtereik a jelentősek, pl. az acetecetészter, amely az α-szénatomján nátrium-etiláttal deprotonálható és az így képződő anion fokozott nukleofilitása sokoldalúan kihasználható. Ez képezi az alapját az acetecetészter szintéziseknek.
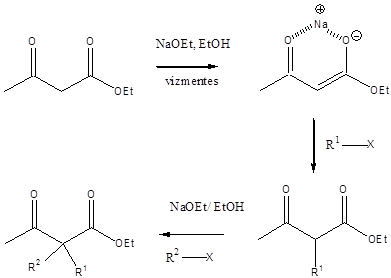
Az így kialakított C-alkilezett acetecetészter kétféleképpen alakítható tovább. A ketonbomlás során az észtercsoportot híg lúggal hidrolizálva, majd a β-oxokarbonsavat melegítéssel dekarboxilezve keton nyerhető. A savbomlás erélyesebb körülmények között tömény lúgot kell használni, melynek során a reakció az oxo-karbonil-csoporton indul, melyet szén-szén kötéshasadás követ, azaz retro-Claisen reakció játszódik le és ecetsav illetve az alkilcsoportokat hordozó észter képződik, melynek hidrolízise a megfelelő karbonsavat adja.
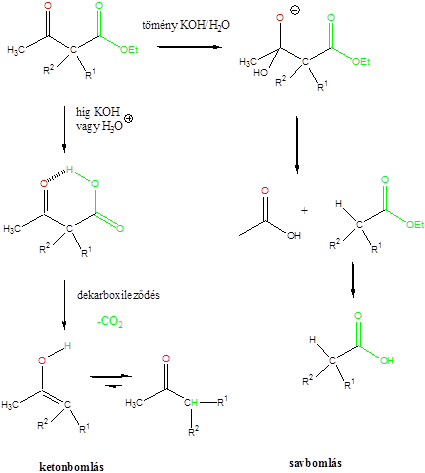
Az acetecetészter- szintézishez hasonlatos a malonészter szintézis. Dietil-malonátból karbaniont létrehozva, majd a metiléncsoportort alkilezve, az észtereket hidrolizálva és savanyítás után enyhe melegítéssel dekarboxilezve α-helyzetben helyettesített karbonsavat kapunk.
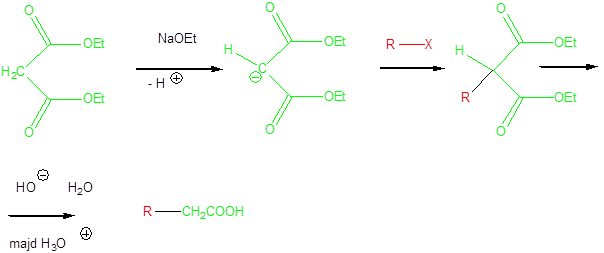
A hangyasav a hangyák (Formica rufa) váladékának alkotója. Erős sav és a benne lévő formilcsoport miatt redukáló szer is.
Az ecetsav jellegzetes szúrós szagú folyadék, vízzel minden arányban elegyedik, vízmentes állapotban 17 ºC-on kristályos tömeggé dermed, ezért jégecetnek is nevezzük.
Az izovaleriánsav a Valeriana officinalis növény gyökerében fordul elő, részben észteresített formában.
A palmitinsav (C16) és a szterainsav (C18) glicerinnel alkotott észterei a szobahőmérsékleten szilárd zsírokban fordulnak elő túlsúlyban, míg a telítetlen zsírsavak inkább az olajokban fordulnak elő. A telítetlen zsírsavakat tartalmazó glicerideket gyakran hidrogénezik nikkel katalizátorral (margarin gyártás), a folyamatot zsírkeményítésnek nevezik.
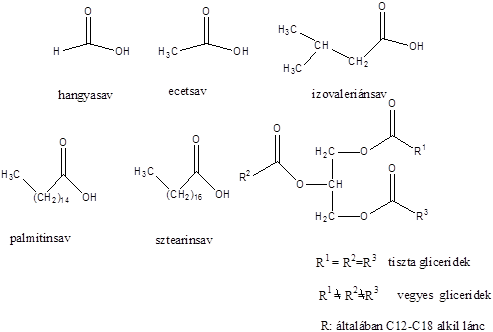
A telítetlen karbonsavak közül az akrilsavat, a cisz kettőskötést tartalmazó oljsavat, a cisz kettőskötéseket tartalmazó linolsavat és linolénsavat valamint az arachidonsavat említjük. A nagy szénatomszámú telítetlen karbonsavak a növényi magvakban és a belőlük préselt olajokban fordulnak elő.
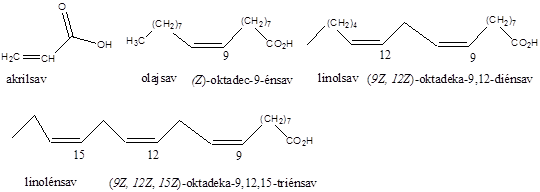
Az archidonsavnak külön élettani jelentősége van, mert az emberi szervezetben a ciklooxigenáz enzimrendszer katalizálta oxidációval endoperoxidon keresztül a vérnyomást és a véralvadást szabályzó prosztaciklinné (PGI2) és tromboxán A2-vé alakul.
A polikarbonsavak közül az oxálsavat említjuk, amely sóska félékben K-só formájában, egyéb növényekben pedig oldhatatlan kalcium-oxalátként fordul elő. Dikarbonsavak közül fontos még a malonsav, a borostyánkősav, a maleinsav (Z-izomer), fumársav (E-izomer). A fumársav és a borostyánkősav a citrátciklusban vesznek részt.
Az aromás karbonsavak közül a benzoesavat említjük meg, melynek nátrium sója az élelmiszeripar fontos konzerváló anyaga. A ftálsav észtereit a műanyagiparban lágyítóként használják. A 2-hidroxibenzoesav triviális neve szalicilsav. A szalicilsav legelterjedtebb származéka az aszpirin (acetilszalicilsav) széles körben alkalmazott láz- és fájdalomcsillapító.
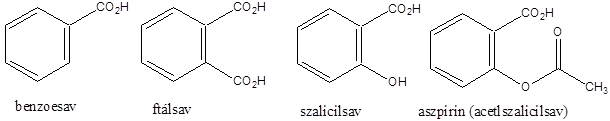
A hidroxikarbonsavak közül említjük a tejsavat, melynek S-enantiomerje az izomszövetben képződik, míg R-enantiomerje a cukrok tejsavas erjedése során keletkezik. Az almasav a legelterjedtebb növényi savak egyike. A citromsav a citrom és sok más gyümölcs levében található. A borkősavnak három sztereoizomerje ismeretes. Ezek közül kettő optikailag aktív, a harmadik a belső szimmetria síkkal rendelkező mezo-borkősav, optikailag inaktív.
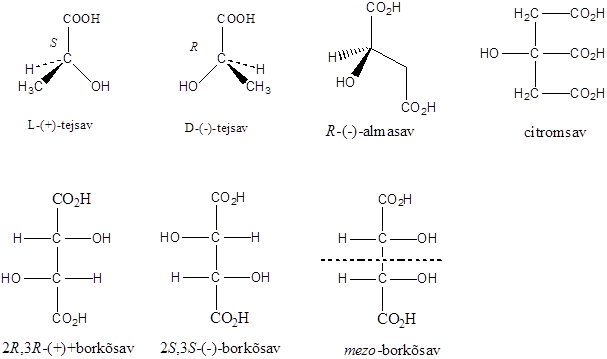
Az oxokarbonsavak közül a piroszőlősav számos biológiai folyamatnak, így a szénhidrátok átalakulásának fontos intermedierje. Az átlakulásokban sokszor az enolformájának foszforsavval alkotott észtereként (foszfoenol-piruvát) vesz részt. Az oxálecetsav szintén biokémiai folyamatokban vesz részt, az almasav enyhe oxidációjával keletkezik.
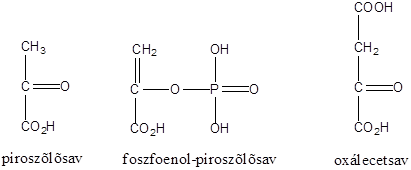
Maga a szénsav labilis vegyület és csak a széndioxid vizes oldatában keletkezik. Az egyensúly a szén-dioxid és a szénsav között a széndioxid irányába tolódik el:
A szénsav sói (karbonátok) már lényegesen stabilabbak, mint a szénsav.
Mivel a szénsavban is megtalálhatók a karbonsavakra jellemző csoportok, a szénsavból előállíthatók, illetve levezethetők a szerves savaknál megismert származékok (savkloridok, savamidok stb.). A szénsav származékai között számos biológiai jelentőségű vegyülettel találkozunk.
- A foszgén a szénsav dikloridja. Szobahőmérsékleten gázhalmazállapotú anyag, erős méreg, az I. világháborúban harcigázként alkalmazták. Oxigén jelenlétében kloroformból napfény hatására is keletkezik. Ammóniával karbamidot képez, etanollal dietil-karbonátot ad.
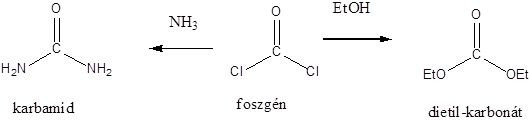
- A karbamidsav a szénsav monoamidja, szabad állapotban nem ismeretes (instabil). Sói a karbamátok és észterei az uretánok már stabil vegyületek. Az ammónium-karbamátot sütőporként alkalmazzák, az uretán (etil-karbamát) a karbamidsav etilésztere.
A karbamid (urea) a szénsav diamidja, az emlősök (köztük az ember) fehérje-anyagcseréjének végterméke. A vizeletben választódik ki; az ember mintegy 25-30 g-ot ürít naponta. Ha a karbamid kiválasztása zavart szenved, a szérum karbamidkoncentrációja, s ezzel a szérum ozmotikus koncentrációja emelkedik. A karbamidot műanyagok (aminoplasztok) előállításához nagy mennyiségben használják fel. A gyógyszeripar barbiturátok és egyéb ureidek előállítására; a vegyipar nitrogéntartalmú műtrágya készítésére használja fel.
A karbamid molekularácsot tartalmazó kristályokat alkot. Ezekben a kristályokban a karbamidmolekulák bal- vagy jobbmenetes csavarvonal mentén helyezkednek el. A molekulákat hidrogénhídkötések rögzítik. A rácsok csatornákat képeznek, amelyekbe megfelelő méretű idegen molekulák is beépülhetnek. Ily módon határozott összetételű zárványvegyületek jönnek létre. Ilyen pl. a hidrogén-hiperoxiddal alkotott zárványvegyület, a hiperol nevű fertőtlenítőszer.
A karbamid salétromossavval, mint minden primer amin, N2-gázt fejleszt:
Hevítéssel ammónia leadása közben biuretté alakul át, ami lúgos közegben Cu2+-ionokkal ibolyaszínű kelátkomplexeket alkot (a peptidek és proteinek biuretreakciója is ezen alapszik):
A karbamid NH2-csoportjai karbonsavak karboxilcsoportjával acilezhetők, a reakció eredményeként savamid jellegű vegyületek, ureidek jönnek létre. Aszerint, hogy a karbamidnak egyik, vagy mindkét aminocsoportja acilezett, mono-, illetve diureideket különböztetünk meg. A diureidek fontos csoportját alkotják a gyűrűs ureidek, amelyeknél a karbamidot kétbázisú karbonsav acilezi. Az ureidek közül jó néhány élettani hatású. Számos nyugtató és altató hatású, gyógyszerként használt vegyület található közöttük. A karbamidnak malonsavval alkotott gyűrűs diureidje a barbitursav.
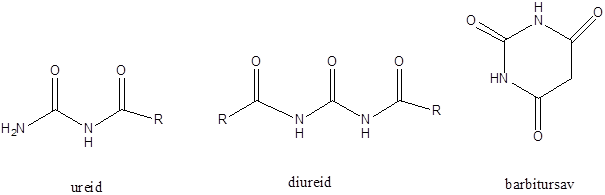
A barbitursav, amely a heterociklus vegyületek közé is sorolható, enol-oxo tautomériát mutat. Számos gyógyászati szempontból jelentős vegyület alapanyaga. Magának a barbitursavnak még nincs altató hatása. Az altató hatású származékok az 5-ös szénatomon két szubsztituenst tartalmaznak (veronal, szevenal). A barbitursavhoz hasonlóan nyerhető 2-tiobarbitursavak származékai, melyek között sebészeti altatószerek találhatók (gyorsan lebomló, kevéssé toxikus intravénás narkotikumok, mint pl. az intranarcon).
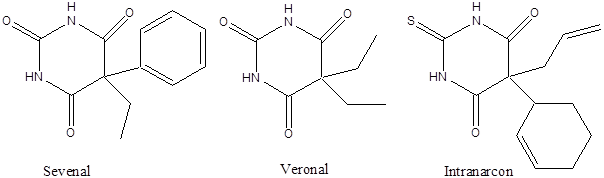
A guanidin a karbamid imidje. Erős szerves bázis, könnyen megköti a protont és a létrejött guanidínium kation szimmetrikus mezomer strukturával (töltés delokalizáció) jellemezhető a legjobban.
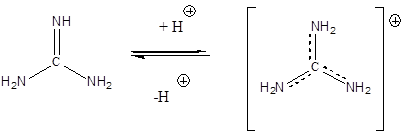
A guanidin számos biológiailag fontos vegyületben megtalálható, így pl. az arginin nevű aminosavban, a kreatinban és a kreatininben (gerincesek izmában, agyában, vérében). A kreatinin a kreatin átalakulási terméke, az emlősök vizeletének egyik alkotórésze.
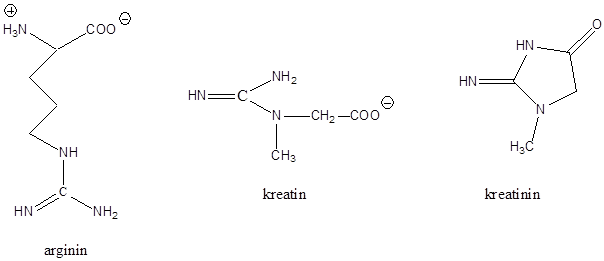
1.) Nevezze el az alábbi karbonsavakat és karbonsav származékokat:
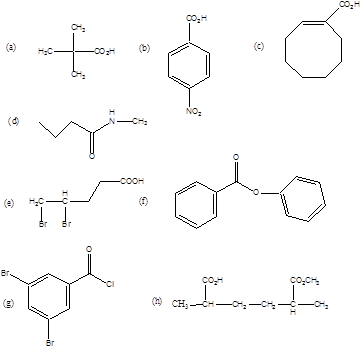
2.) Az alábbi sorozatokban állítsa sorba a karbonsavakat erősségük szerint (a legerősebb legyen az első):
(a)ecetsav, trifluórecetsav, klórecetsav
(b)benzoesav, szalicilsav, 4-nitrobenzoesav (c) hangyasav, oxálsav, hexándisav (adipinsav)
3.) Az 5-hidroxipentánsav savas közegben egy ciklusos vegyületet ad. Rajzolja fel a szerkezetét!
4.) Mi lesz az alábbi reakciók terméke?
5.) Az alábbi vegyületek hogyan szintetizálhatók vajsavból?
6.) A brómecetsav disszociációs állandója Ka = 1 x 10-3 . Számítsa ki a 0,1 M vizes brómecetsav oldat pH-ját!
7.) A kokaint lúgos hidrolízisnek vetjük alá. Mi lesz a hidrolízis terméke (3 vegyület)?
8.) Az N,N-dietil-3-metilbenzoesavamidot gyakran használják szúnyog, légy és kullancs riasztó krémek adalékanyagaként. Hogyan szintetizálná a vegyületet 3-brómtoluolból kiindulva?
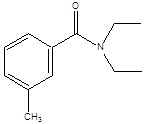
9.) Az alábbi vegyületek közül melyik állítható elő malonészter szintézissel?
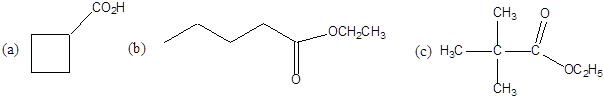
10.) Adja meg a szerkezetét az-5-etil-5-(pent-2-il)-2-tiobarbitursavnak. Adja meg a szintézishez szükséges reaktánsok szerkezetét is.
Tartalom
- 32. Aminosavak, peptidek, fehérjék csoportosítása
- 33. α-Aminosavak fizikai tulajdonságai és sav-bázis jellege
- 34. Az α-aminosavak térszerkezete
- 35. Az α-aminosavak előállítása
- 36. Az α-aminosavak reakciói
- 37. Peptidek és fehérjék szerkezete
- 38. A fehérjék csoportosítása
- 39. Peptidek szintézise
- 40. Feladatok
Az aminosavak olyan helyettesített karbonsavak, amelyekben a szénhidrogéncsoportok egy vagy több hidrogénjét aminocsoport helyettesít. Az aminosavakat a szénhidrogénrész jellege (alifás/aromás), valamint az aminocsoportnak a karboxilcsoporthoz viszonyított helyzete szerint osztályozzuk, és így megkülönböztethetünk α-, β-, γ- stb. aminosavakat, illetve aromás aminosavakat.
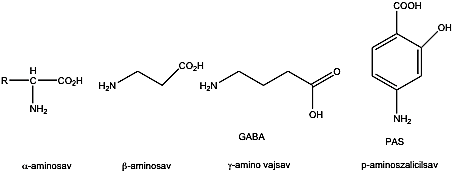
A különféle típusú aminosavak közül legjelentősebbek az α-aminosavak, mert ezekből épülnek fel az élő sejt anyagállományának nélkülözhetetlen alkotórészei, a fehérjék. Sokféle fehérjét ismerünk, így a fehérje elnevezés gyűjtőfogalom, hasonlóan a szénhidrogénekhez. A közismert tojásfehérje is sok száz különböző fehérje vizes oldata, melyből körülményes munkával tiszta, egységes fehérjéket lehet elkülöníteni. Ezek teljes hidrolízisénél α-aminosavak, részleges hidrolízosénél pedig peptidek keletkeznek. A peptidek két vagy több α-aminosavból (továbbiakban aminosavból) peptidkötéssel felépülő molekulák. A peptidkötés olyan savamidkötés, amely az egyik aminosav karboxilcsoportja és a másik aminosav α-aminocsoportja között alakul ki. Minden egyes savamidkötés létrejötte egy molekula víz lehasadásával jár, vagyis a peptidek és a fehérjék az aminosavak polikondenzációs termékei.
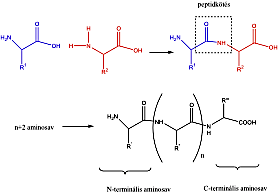
A fehérjék kémiai szempontból két csoportba sorolhatók. Az egyszerű fehérjék a proteinek (csak aminosav részekből állnak), az összetett fehérjék, a proteidek (az aminosav részeken kívül még más alkotórészt is tartalmaznak). Az egyszerű fehérjék több száz aminosavrészt tartalmaznak. Általában, ha az aminosav részek száma kevesebb mint 100, akkor nem fehérjékről, hanem polipeptidekről vagy peptidekről beszélünk. A peptideket az aminosav részek száma szerint csoportosítva megkülönböztetünk di-, tri-, tetra-, stb. peptideket.
A fehérjék és a peptidek peptidláncának egyik végén aminocsoport, a másikon karboxilcsoport van. Az előbbit N-, a másikat C-terminális láncvégnek nevezzük. A peptidlánc szokásos felírása szerint az N-terminális láncvég bal oldalon van és jobbra folytatódik a lánc. A fehérjék és peptidek teljes hidrolízisével mintegy 20-féle aminosav nyerhető, közöttük vannak monoamino-monokarbonsavak, továbbá második bázisos (amino- vagy guanidino-) vagy egyéb (hidroxil-, szulfhidril-) csoportot is tartalmazó aminokarbonsavak, valamint izo- vagy heterociklusos szerkezeti részeket is magukba foglaló aminokarbonsavak. Különleges szerkezetű hidrolízistermékek a prolin és a hidroxiprolin, melyek α-helyzetben gyűrűbe zárt, bázisos jellegű iminocsoportot tartalmaznak. Az aminosavak másik megkülönböztetési módja, hogy esszenciálisak-e vagy sem. Előbbieket táplálék formában kell felvennünk, az alábbiakban a név mellett „E”-vel jelöltük.
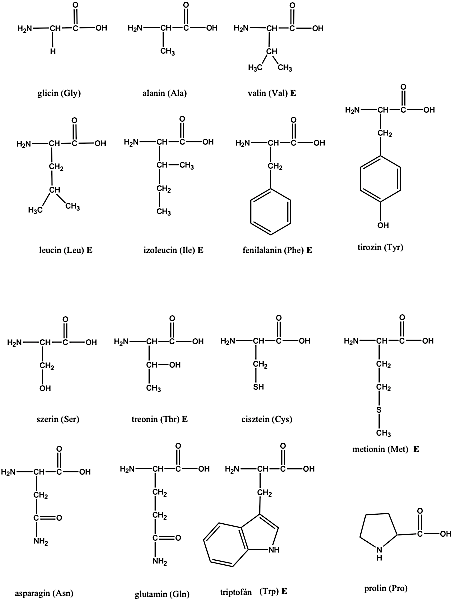
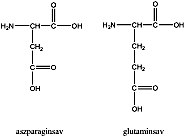
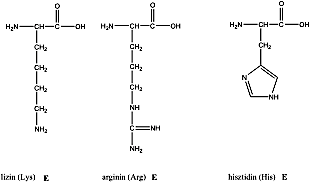
Az α-aminosavak kristályos, magas olvadáspontú vegyületek. Olvadáspontjuk sokkal magasabb, mint azoké a karbonsavaké vagy aminoké, melyekből helyettesítéssel levezethetők. Olvadáspontjuk fölött elbomlanak, gázhalmazállapotban nem létképesek. Oldékonyságuk is a sókra emlékeztet. Szerves oldószerekben, például alkoholban a prolin és a hidroxiprolin kivételével gyakorlatilag oldhatatlanok, míg vízben valamennyi jól oldódik. Oldatban, az oldat pH-jától függően kationként, anionként vagy ún. ikerionos alakban vannak jelen. Minthogy a vegyületek egyidejűleg bázikusak és savasak, a savas csoport (-COOH) átadja a protonját a bázisosnak (-NH2), és így keletkezik az ikerionos forma.
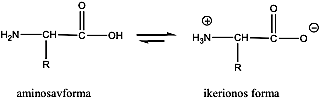
Az aminosavak ikerionos szerkezetű molekuláinak ugyanannyi a pozitív töltése, mint a negatív. A molekulák „bruttó” töltése tehát nulla, azaz vizes oldatban izoelektromos állapotban vannak. Nyilvánvaló, hogy vizes oldatban a következő sav-bázis egyensúlyi rendszer jelenlétével kell számolnunk.
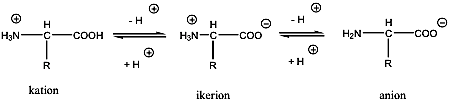
Az egyensúly helyzetét a vizes oldat hidroxóniumion-koncentrációja (pH-értéke) szabja meg. Ha a vizes oldathoz erős savat (pl. sósavat) adunk, akkor a molekula gyengén bázisos-anion jellegű – karboxilcsoportja is protonálódik és átalakul kationná, azaz az aminosav hidrokloridja (só) keletkezik. Erős bázis (pl. NaOH) hozzáadásakor, a hidroxidionok az aminosav ammóniumcsoportjáról leszakítják a protont, és a molekulát anionná alakítják át, melynek negatív töltését a lúgból visszamaradt nátriumion semlegesíti. Éppen ezért az aminosavak lúgos oldatuk elektrolízisekor mint anionok az anód felé, míg savas oldatuk elektrolízisénél kationként a katód felé vándorolnak. Azt az állapotot, amelyre az jellemző, hogy az oldott aminosav az egyenáram hatására nem mozdul el sem a katód, sem az anód irányába, izoelektromos állapotnak, a hozzá tartozó pH-értéket izoelektromos pontnak (pI) nevezzük. Ez az érték az aminosavra jellemző fizikai állandó.
α-Aminosavak molekulái – a glicin kivételével – királisak, és ennek megfelelően két tükörképi konfigurációba rendezhetők, melyeket a Fischer-féle jelölést használva L-, illetve D-betűkkel különböztethetünk meg. A természetben előforduló aminosavak túlnyomó többsége az L-sorban és (S)-abszolút konfigurációjú, mint azt az R-(+)-glicerin-aldehiddel történő kémiai korrelációjuk, valamint röntgenvizsgálataik is igazolták. Az L-cisztein esetén a C.I.P. szabály alapján a prioritási sorrend miatt nem az (S)-, hanem az R-konfiguráció adódik.
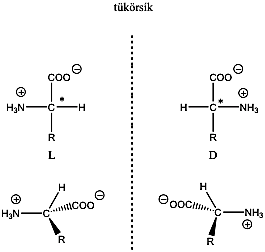
Az α-halogénezett savak ammónium hidroxiddal aminosavakká alakíthatók át.
Aldehidek ammóniával aldimint szolgáltatanak vízeliminációt követően. Az aldiminre történő hidrogén–cianid addíció α-aminonitrilt ad, ami aminosavvá hidrolizálható (Strecker-Zelinszkij-féle szintézis). A reakciót kényelmesebb ammónium-klorid és nátrium cianid együttes használatával kivitelezni.
Tartalom
Az aminosavak, hasonlóan az aminokhoz, alkilezőszerekkel (pl. dimetil-szulfáttal) lúgos közegben negyedrendű aminokká, ún. betainokká alakíthatók.
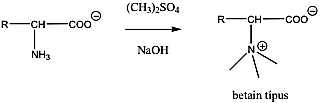
Az aminosavak savmegkötő jelenlétében acilezhetők. A klórhangyasav-benzilészterrel való acilezés különösen a peptidszintézisekben jelentős, danzil-kloriddal pedig fluoreszkálóvá tesszük az aminosavat, amely az optikai detektálását teszi lehetővé (pl. kromofor csoporttal nem rendelkező aminosav esetén).
A primer aminokhoz hasonlóan az aminosavak már szobahőmérsékleten reagálnak salétromossavval és pillanatszerűen a megfelelő hidroxisav és nitrogén képződik.
Oxidáció: az élő szervezetben fontos szerepet játszik az aminosavak oxidálása α-iminosavakká, melyek vizes közegben α-ketosavakká és ammóniává hidrolizálnak. A szervezetben ezt az oxidációt enzimek végzik, in vitro pedig hidrogén-peroxiddal végezhető el.
Az α-aminosavak ninhidrinnel reagálva lila színű vegyületet adnak. Ezt használták régen ujjlenyomatok rögzítésére.
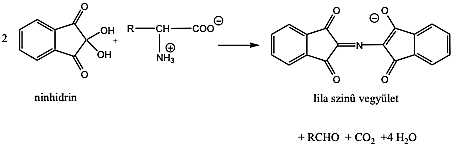
Az aminosavak karboxilcsoportjai a Fischer-féle módszerrel észteresíthetők, melynek során az aminosavészter kristályos hidrokloridját kapjuk, ebből az aminosav-észtert bázissal szabadíthatjuk fel.
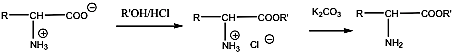
Az aminosavak nehézfém-ionokkal (pl Cu(II)-val) vízben nagyon rosszul oldódó komplex sókat képeznek, melyekből az aminosav savas közegben kén-hidrogénnel szabadítható fel:
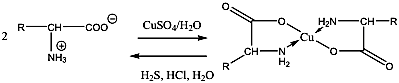
A legelső peptidszintézis azon a megfigyelésen alapszik, hogy az aminosavészterek alkohol kilépése közben diketopiperazin származékokká alakulnak, amelyek savval vagy lúggal dipeptiddé alakíthatók.
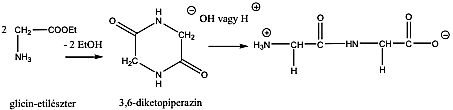
Tartalom
A különböző fehérjék sósavas hidrolízise – a korábbiak szerint – húszféle aminosavat eredményez, amelyek a fehérjékben peptidkötéssel (amidkötés) kapcsolódnak össze.
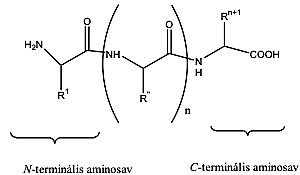
Már említettük, hogy a fehérje vagy polipeptid lánczáró részei balról jobbra haladva az N- és C-terminális aminosavegységek (terminus latin szó, határt, valaminek a végét jelenti). Az aminosavak sorrendjét az N-terminálistól a C-terminális felé haladva aminosavszekvenciának nevezzük. Az aminosavszekvencia a fehérjék elsődleges szerkezete. Az N-terminális aminosavat a például Edman-lebontással határozhatjuk meg. Az Edman-lebontás során a fehérjét vagy a peptidet fenil-izotiocianáttal reagáltatjuk, majd a keletkezett fenil-tiokarbamid-származékból (PTC-peptid) vizes sósav hatására 5-helyzetben helyettesített fenilhidantoin (PTH) hasad le, amelynek szerkezetmeghatározásával az N-terminális aminosav azonosítható. A lebontás n számú ismétlésével az aminosavak kapcsolódási sorrendje meghatározható.
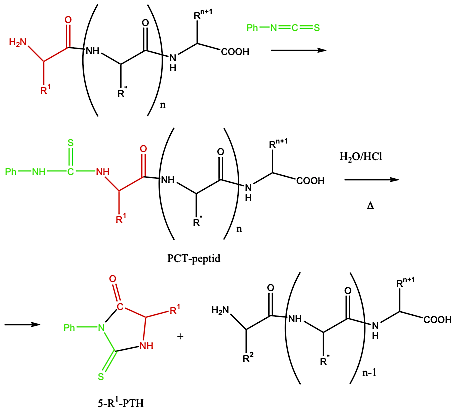
A másodlagos szerkezet alatt a helyi konformációt értjük, ami főleg α-hélix vagy β-redőzött szerkezet. A polipeptidlánc konformácios viszonyainak kialakításában nagy szerepet játszanak az amidrészletek konformációs viszonyai is. Az amidcsoportban a karbonilszénatom és a nitrogénatom közötti konjugáció miatt a C-N kötés körül gyakorlatilag nincs forgás, azaz a síkalkatú amidcsoport meglehetősen merev.
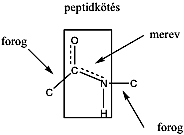
A polipeptidláncok leggyakoribb másodlagos szerkezete a csigavonalban feltekeredő stuktúra, az α-hélix. Termodinamikailag a jobbmenetes (az óramutató járásával megegyező) α-hélix a legstabilabb. Ebben a szerkezetben az aminosav oldalláncok a hengernek tekinthető burkolófelület külső részén helyezkednek el. Egy fordulatra 3,6 aminosav jut, a menetemelkedés nagysága 0,54 nm. Az α-hélixet maximális számú hidrogénkötés rögzíti.
A teljesen nyújtott β-redőzött lemez szerkezetben polipeptidláncok, azonos (paralell) vagy ellentétes (antiparalell) irányultsággal az N-terminálistól a C-terminális irányába, egymással párhuzamosan helyezkednek el. A teljesen nyújtott szerkezet nem kedvezményezett, ezért a Cα-atomoknál a láncok megtörnek és harmonikaszerű, cikk-cakk alakzatot vesznek fel.
β-szerkezethez sorolják a β-hajtű szerkezetet is. A β-hajtű általában 4 aminosavból álló kis peptidszakasz, amely az antiparalell β-redőzőtt szerkezeteket, vagy a β-redőket és az α-hélixeket köti össze.
A meghatározott másodlagos szerkezetű polipeptidláncok a térben egymással kölcsönhatásba lépve alakítják ki a háromdimenziós térszerkezetet, amelyeket harmadlagos szerkezetnek hívunk. A harmadlagos szerkezetnek két jellegzetes típusa a fibrilláris (fonalszerű) és a globuláris (gömbszerű) fehérjeszerkezet. A fibrilláris fehérjék esetén a polipeptidláncok egymással párhuzamosan elrendeződve szálas vagy réteges szerkezetet alakítanak ki. Az ilyen fehérjék nehezen oldhatók, biológiai funkciójukat tekintve védő, elválasztó hártyákat, mechanikai munkát végző, rostos szerkezetű rendszereket alakítanak ki (haj, köröm, izom stb.). A legegyszerűbb szerkezetben a ß-láncok hidrogénkötésekkel kapcsolódva paralell és antiparalell lefutású szálakat hoznak létre. Fibrilláris fehérjék hélixes polipeptidláncokból is kialakulhatnak, mint például a keratin (bőr, haj) és a kollagén (kötőszövet). A kollagénben három balmenetű hélix alkot egy szuperhélixet. A globuláris fehérjék térbeli szerkezetében a hélixes formák mellett lemezes szerkezeti részek is előfordulnak. Biológiai funkciókat tekintve a globuláris fehérjék főként enzimek.
A polipeptidláncot alkotó aminosavak oldalláncaiban lévő csoportok megfelelő térbeli helyzetben másodlagos kötések kialakítására képesek mind a polipeptidláncon belül, mind több polipeptidlánc között.
A legjellemzőbb másodlagos kötések:
1. A diszulfidkötések amelyek a ciszteinek szulfanilcsoportjainak összekapcsolódása révén jöhetnek létre
2. a hidrogénkötések, amelyek mindenütt létrejöhetnek, ahol a hidrogének elektronegatív atomok közelébe kerülnek
3. a hidrofób csoportok kölcsönhatása révén kialakuló kapcsolatok
4. ionos kötések, amelyek a protonált aminocsoportok és a negatív töltésű karboxilcsoportok között alakulnak ki
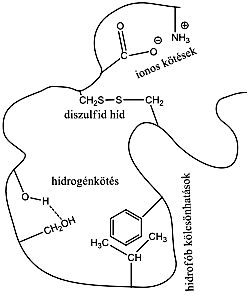
A másodlagos és a harmadlagos szerkezetet együttesen a fehérjemolekula konformációjának nevezzük.
A főként globuláris fehérjékből képződő, másodlagos biológiai funkcióval rendelkező egységek alakítják ki a negyedleges szerkezetet. Az asszociáló polipeptidláncok lehetnek azonos vagy eltérő kémiai felépítésűek. A keletkező oligomer szerkezeti elemeit a görög ábécé betűivel jelöljük, alsó indexben a polipeptidláncok számának megadásával. Az alábbi ábrán a hemoglobin negyedleges szerkezetét láthatjuk, amely 4 alegységből α1, α2, β1, β2 tevődik össze.
- Biológiai funkció szerint
A fehérjék csoportosítása sokféle szempont szerint történhet. Az egyik leglényegesebb a biológiai funkció szerinti osztályozás: pl. enzimek, hormonok, transzport fehérjék, vázfehérjék.
- Molekulaalkat szerint
Egy másik felosztás a fehérjéket alkatuk, illetve alakjuk alapján csoportosítja, fibrózus és globuláris fehérjéket különböztetve meg egymástól.
A fibrózus fehérjékhosszú, fonalszerű molekulák. Ebbe a csoportba tartoznak a támasztó- és kötőszöveti fehérjék, mint pl. a kollagén, fibroin, elasztin, keratin, mukoproteinek.
A globuláris fehérjék tömörebb, ellipszoin vagy gömb alakú molekulák. Ide tartoznak a szérumfehérjék, a tojásfehérje, az emzimek nagy része stb.
- Oldhatóság alapján
A fehérjék további felosztási módja az oldhatóságukon alapuló csoportosítás. Oldhatóságuk alapján az alábbi főbb csoportokat különböztetjük meg:
Albuminok
Desztillált vízben jól oldódnak az albuminok. Oldatukból csak tömény (70%-os telítettségű) ammonium-szulfáttal csaphatók ki. Az élő szervezetben széles körben elterjedtek: pl. a vérben lévő szérumalbumin, a tejben lévő laktalbumin, a tojásban lévő ovalbumin stb.
Globulinok
Desztillált vízben nem, de híg sóoldatban jól oldódnak a globulinok. Az albuminoktól ammónium-szulfátos frakcionálással különíthetők el, ti. a globulinok már 0,5 % telítésű ammoniumszulfát-oldatban kicsapódnak, amiben az albuminok még oldatban maradnak. A globulinok jellegzetes képviselői: a fibrinogén és a belőle véralvadáskor képződő fibrin, a vérszérum α-, ß- és γ-globulinja, az izomban található miozin és aktin. A szérum γ-globulinját a gyógyászatban is felhasználják.
Hisztonok és protaminok
Az ebbe a csoportba tartozó fehérjék csak savas közegben oldódnak, vizes oldatukból ammóniával kicsaphatók. Sok arginint tartalmazó, bázikus fehérjék. Hődenaturációval szemben viszonylag ellenállók. Sejtmagokban nukleinsavakhoz kötve fordulnak elő.
Prolaminok éss gliadinok
A prolaminok és gliadinok 50-90%-os alkoholban oldódó fehérjék. Általában jelentős mennyiségű prolint, illetve glutaminsavat tartalmaznak. Főleg növényi magvakban fordulnak elő (a liszt legfontosabb fehérjekomponensei, mint pl. a sikér, ide tartoznak).
Vázfehérjék
A szkleroproteinek vagy vázfehérjék vízben és más oldószerben nem oldódnak. A kötőszövetben találhatók (kollagén, keratin, elasztin stb.).
- Egyszerű és összetett fehérjék
Csoportosíthatók a fehérjék összetételük alapján is. Eszerint megkülönböztetünk egyszerű és összetett fehérjéket (proteineket és proteideket).
Az egyszerű fehérjék hidrolízise során gyakorlatilag csak aminosavak keletkeznek, az összetett fehérjék viszont olyan komplexek, amelyek proteinrészből és nem fehérje természetű, ún. prosztetikus csoportból épülnek fel. Hidrolízisük alkalmával az aminosavak mellett más jellegű vegyületek (szénhidrátok, lipidek, nukleinsavak, színes festékjellegű anyagok, fémek, foszfát) is hasadnak ki.
Az összetett fehérjékben lévő prosztetikus csoport alapján megkülönböztetünk kromoproteineket, amelyek színes prosztetikus csoportot tartalmaznak (pl. hemoglobin, klorofill, látóbíbor), foszforproteineket (pl. kazein), glikoproteineket (pl. a szénhidrátot tartalmazó heparin,γ-globulin), lipoproteineket és nukleoproteineket.
A peptidszintézisekben az aminosavak közötti amidkötés kialakítása az ún. oldat- és szilárd- fázisú technika segítségével valósítható meg. Az oldatfázisú eljárásoknál az N-terminális aminosav aminocsoportját védőcsoporttal látják el, majd a karboxilcsoportot aktiválják, és végül az így nyert védett és aktivált aminosavat reagáltatják a C-terminális aminosavval.
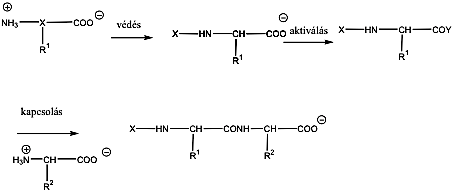
A kapott dipeptid karboxilcsoportjának aktiválását követően a kapcsolás elvileg tetszés szerinti számban megismételhető.
A védőfeladatot leggyakrabban benzioxikarbonil (Cbz)- vagy terc-butoxikarbonil (Boc)- vagy (9-fluorenilmetikloxi)-karbonil (Fmoc)-csoport látja el, mivel ezek eltávolítása racemizáció veszélye nélkül enyhe körülmények között elvégezhető.
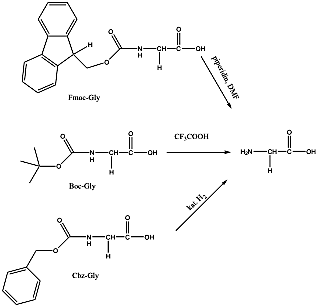
A karboxilcsoport aktiválása savklorid-, savazid-, aktív észter- és vegyes anhidridcsoporttá történő átalakításuk útján érhető el. Aminocsoportjukon védett (pl. Cbz- vagy Boc-csoporttal) aminosavak diciklohexilkarbodiimid (DCC) jelenlétében közvetlenül is kapcsolhatók aminosavészterekkel (pl. benzilészterrel). A reakció első lépésében az aminocsoportján védett aminosav a DCC-vel reagálva a megfelelő O-acilezett izokarbamidszármazékká alakul, amely a karbonilcsoportján kialakuló csökkent elektronsűrűség miatt készségesen reagál az aminosavészter aminocsoportjával. Az észtercsoportnak karboxilcsoporttá történő alakítása (pl. kat./H2) után a kapcsolás további aminocsoportján védett dipeptiddel megismételhető.
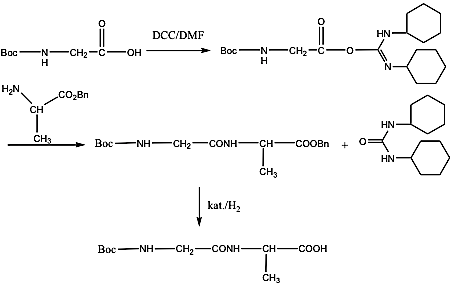
A DCC-t használják a peptidkötés kialakítására a Merrifield (Nobel-díj, 1984) által kidolgozott szilárd fázisú peptiszintézis során is. E módszernél a C-terminális aminosavat olyan divinilbenzollal térhálósított polisztirol polimerhez kötik, amelyek körülbelül minden századik fenilcsoportja klórmetilcsoportot tartalmaz. Az így rögzített aminosavhoz DCC-vel, aminocsoportján Boc- vagy Fmoc-csoporttal védett aminosavakat kapcsolnak.
A kapcsolási lépés a polimerhez kötött peptiden a védőcsoport eltávolítása után megismételhető. A módszer előnye, hogy a szennyezések és melléktermékek a polimerből könnyen kimoshatók és az eljárás automatizálható. A szintézis utolsó lépéseként a peptid a polimerről vízmentes hidrogén-fluoriddal hasítható le.
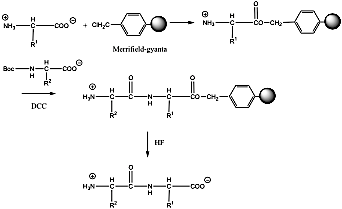
1.) Melyik az a két aminosav amely egynél több kiralitáscentrumot tartalmaz?
2.) Rajzolja fel a lizin és az aszparaginsav domináns formáit pH = 3 és pH =9 kémhatású vizes oldatokban.
3.) Mi lesz a reakciótermék ha a valint az alábbi reagensekkel reagáltatjuk?
a) EtOH/HCl b) ninhidrin c) NaNO2 /HCl d) NaOH
e) d) terméke + benzoszulfonsav-klorid f.) NaOH és dimetil-szulfát
4.) Rajzolja fel a képződő fenilhidantoinok szerkezetét ha Edmann-lebontással az alábbi tripeptid szerkezetét határozzuk meg: Val-Leu-Cys
5.) Mi lehet a minimális molekulasúlya annak a fehérjének, amely 0,29% triptofánt tartalmaz?
6.) Az „aszpartám” nevezetű édesítőszer egy dipeptid metilésztere: Asp-Phe-OCH3. Rajzolja fel az aszpartám szerkezetét. Az aszpartám izoelektromos pontja 5,9. Rajzolja fel az aszartám szerkezetét 7,6-os pH-n.
7.) Egy hexapeptid összetétele a következő: Arg, Gly, Leu, Pro3 és az és a C-terminálison prolint tartalmaz. Mi a hexapeptid aminosav sorrendje, ha hidrolízisekor az alábbi fragmenseket kaptuk: Gly-Pro-Arg, Arg-Pro, Pro-Leu-Gly?
Tartalom
- 41. Öttagú heterociklus vegyületek
- 42. Csoportosítás:
- 43. Nevezéktan:
- 44. Öttagú, egy heteroatomot tartalmazó heterociklusok
- 45. Öttagú, több heteroatomot tartalmazó vegyületek:
- 46. Hattagú, egy heteroatomot tartalmazó vegyületek
- 47. Nukleotidok, nukleinsavak
- 48. Feladatok a heterociklusok és nukleinsavak köréből:
Azokat a gyűrűs vegyületeket, amelyek a gyűrűben szénatomok mellett heteroatomot (O, N, S, stb.) is tartalmaznak, heterociklusos vegyületeknek nevezzük.
-A gyűrű tagszáma szerint
-A heteroatomok minősége szerint
-A heteroatomok száma szerint
A telítetlen heterociklusos vegyületek jelentős része aromás sajátságokkal rendelkezik. Ezek az ún. heteroaromás vegyületek. Ebben a fejezetben ezekkel a heteroaromás vegyületekkel, illetve részlegesen telített származékaikkal fogunk foglalkozni. Azon belül is a legelterjedtebb és legnagyobb jelentőséggel bíró oxigén, kén, nitrogén tartalmú öt- és hattagú származékokkal.
Tartalom
A monociklusos alapváz nevét a heteroatom nevéből származó előtag(ok)ból pl.: oxa-(O), tia-(S), aza-(N) és a az alábbi táblázatban feltüntetett szótövekből képezzük. A heteroatomokat a oxa-, tia-, aza- sorrendben kell a név elején felsorolni, és szükség esetén a heteroatomok helyzetét a név elején közölt helyzetszámokkal jelezzük. A gyűrűt a legrangosabb heteroatomon kezdve úgy számozzuk meg, hogy a heteroatomok a lehető legkisebb helyzetszámokat kapják. A telítetlenség mértékét vagy a szótőben tüntetjük fel (ld. az alábbi táblázatot), vagy a dihidro-, tetrahidro-, stb. előtaggal jelezzük, ha a heterociklus az alapnévben jelzett maximálisnál kevesebb számú nem kumulált kettős kötést tartalmaz.
Heterociklusos vegyületek nevében a gyűrűre utaló szótövek:
43.1. táblázat -
|
gyűtagszám |
telítetlen |
telített |
|
3 |
irén |
irán* |
|
4 |
et |
etán* |
|
5 |
ol |
olán* |
|
6 (O, S) |
in |
án |
|
6 (N) |
in |
inán |
*A N-tartalmú gyűrűkre az „iridin”, „etidin”, „olidin” végződést előnyben kell részesíteni.
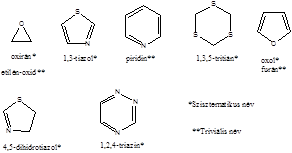
Tartalom
A három- és négytagú egy és több heteroatomos heterociklusokkal nem foglalkozunk. Ezeknek általában kisebb a jelentőségük, mint az öt-, illetve hattagú heterociklusos vegyületeknek. Kiemelkedik azonban közülük a négytagú, egy nitrogént tartalmazó telített heterociklus, az azetidin, ennek is a 2-oxo-származéka, az azetidin-2-on. Ez az ún. -laktám számos természetes és félszintetikus antibiotikumnak fontos szerkezeti eleme.
Az öttagú egy heteroatomot tartalmazó alapheterociklusok:
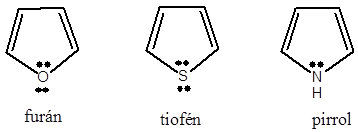
Mindhárom vegyületre teljesül az aromás rendszer kialakulásának valamennyi feltétele:
-sp2 hibridállapotú atomok, így koplanáris a szerkezet
- valamennyi atomnak van pz orbitálja
-a pz elektronok száma 6 (4n+2)
A delokalizációs energiák furán (68 kJ/mol), pirrol (90 kJ/mol), tiofén (122 kJ/mol) kisebbek, mint a benzol (150 kJ/ mol) delokalizációs energiái, ennek oka, hogy a heteroaromás rendszer nem szimmetrikus.
Mindhárom heterociklusban a heteroatomok elektronegatívabbak a szénnél. A –I effektus és az +M effektus eredője határozza meg a tényleges elektronelosztást. Mivel X=NH esetében +M>-I, X= O vagy S esetében +M<-I, ezért alapállapotban a negatív töltéssűrűség a pirrol esetében a szénatomokon, a furán és a tiofén esetében a heteroatomon nagyobb. Ezt a dipólus momentum iránya és nagysága is tükrözi.
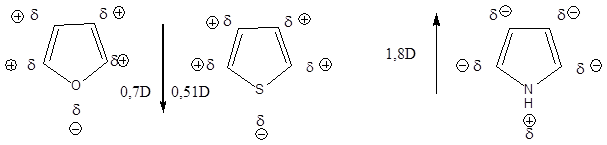
A polarizálhatóság sorrendje: +Epirrol> +Efurán> +Etiofén >> Ebenzol. A három vegyület aromás elektrofil szubsztitúciós készsége nagyobb mint a benzolé, és a sorrend tiofén< furán<pirrol. A furán aromaticitása kisebb, ennek megfelelően előtérbe kerülnek addíciós reakciók is.
1,4-dikarbonil vegyületekből, Paal-Knorr-szintézissel:
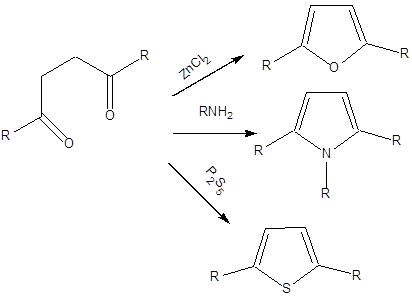
A furán alapvázát legegyszerűbben nyálkasavból állíthatjuk elő:
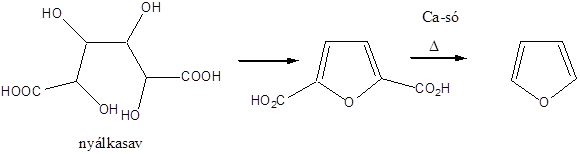
A nitrogén nemkötő elektronpárja részt vesz a 6π-elektronrendszer kialakításában, ezért protont nem képes megkötni, sőt NH-savnak tekinthető, káliummal sót képez.
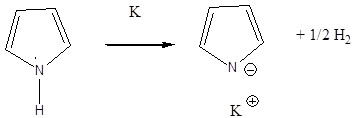
A furán, pirrol, tiofén reakciókészsége nagymértékben emlékeztet a fenol és az anilin elektrofilekkel szembeni reakciókészségére. A furán és a pirrol lényegesen reakcióképesebb a tiofénnél. A szubsztitúció elsősorban a 2-es helyzetben játszódik le, mivel az így képződő π-komplex stabilisabb (rezonancia stabilizáltabb) mint a 3-as szubsztitúcióhoz vezető π-komplex.
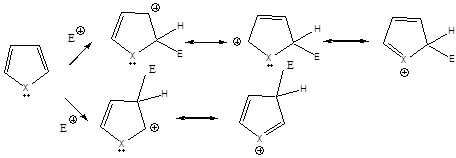
Mindhárom heterociklus hevesen reagál. Elemi brómmal a furán olyan hevesen reagál, hogy tiszta termék nem izolálható.
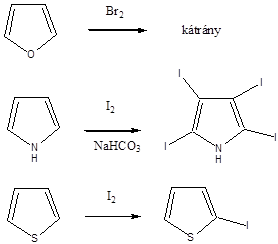
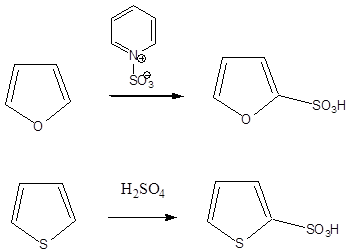
Salétromsav-kénsav elegy hatására mindhárom heterociklus elbomlik, acetil-nitráttal viszont a furán AdE+E, a tiofén viszont SE reakcióban nitrálható.
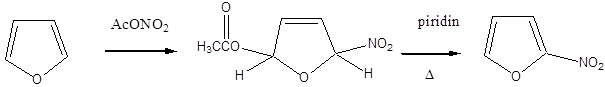
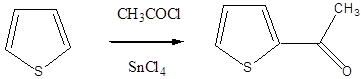
A pirrol króm(VI)-oxiddal maleinimiddé oxidálható. A tiofén peroxisavakkal a már nem aromás szulfonná oxidálódik, amely Diels-Alder-típusú reakcióban dimerizál.
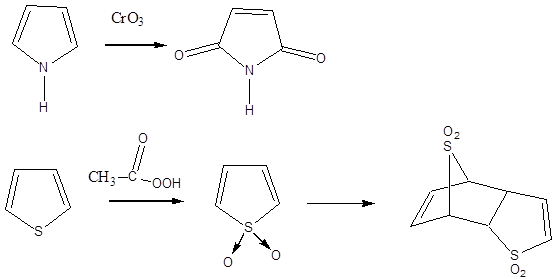
A legkisebb delokalizációs energiájú furán a maleinsav-anhidriddel mint dién reagál.
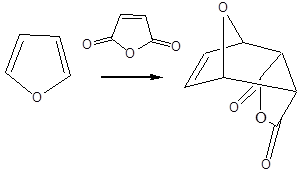
A pirrol benzokondenzált származéka a biológiailag igen fontos indol. Szintézise történhet a Fischer-féle indol szintézissel fenil-hidrazinból és aktivált metiléncsoportot tartalmazó ketonokból sav ill. Lewis-sav jelenlétében:
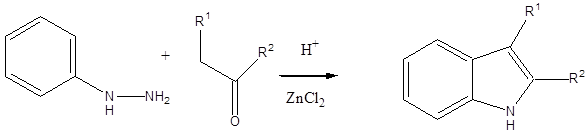
Az indol az öttagú gyűrűjén szintén készségesen lép reakcióba, azonban a szubsztitúció a 3-as helyzetben következik be. Brómozáskor 3-brómindol, Mannich-reakció során pedig gramin keletkezik.
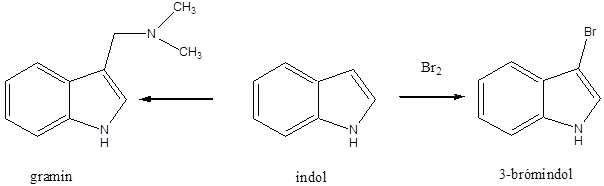
Tetrahidrofurán (THF), fp.: 65 °C. Előállítása: a furán erélyes körülmények közötti katalitikus hidrogénezésével, vagy bután-1,4-diolból. Alifás éter típusú vegyület, Grignard-reakciók, fémhidrides redukciók és alkil-lítiumos reakciók oldószere. Szintézis alapanyaga a bután-1,4-diolnak, a 4-klórbutanolnak, a tetrametilén-dibromidnak (ezen keresztül a nylon 66-nak).
Prolin, hidroxiprolin (pirrolidin-2-karbonsav, 4-hidroxipirrolidin-2-karbonsav): állati fehérjékben előforduló -aminosavak. A természetben az (S)-konfigurációjú fordul elő.
A porfinváz négy, metincsoportokkal összekapcsolt pirrolgyűrűből álló makrociklus, melyben valamennyi nitrogén- és szénatom folytonosan konjugált rendszerben helyezkedik el. A hemin ( a hemoglobin nem fehérje komponense) és a klolofill (a növényi sejtek kloroplasztiszainak alkotóeleme) a biológiai oxidációban, ill. a szén-dioxid asszimilációban döntő szerepet játszó vegyületek.
A hemin és a klorofill abban különböznek egymástól, a pirrolgyűrűkön eltérő szubsztituensek találhatók. A hemin koordinatíve Fe2+ iont köt meg, míg a klorofill Mg2+ iont.
A triptofán indolvázat tartalmazó, fehérjékben előforduló -aminosav.
Triptamin a triptofán dekarboxilezésével nyerhető, fontos szintézisek alapanyaga. A triptamin származéka a szerotonin (5-hidroxitriptamin), ami fontos szerepet játszik az idegsejtek információcseréjében. A melatonin (N-acetil-5-metoxitriptamin) az alkalmazkodást segítő hormon hatású vegyület („alvási hormon”).
Indolvázat tartalmazó természetes, majd mesterségesen is előállított csávaszinezék az indigó.
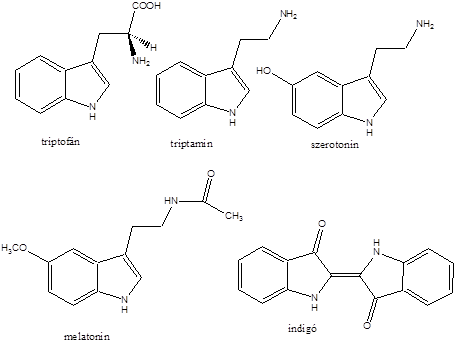
Tartalom
A két vagy több heteroatomos öttagú, maximális számú nem kumulált kettős kötést tartalmazó heterociklusok közül azok aromásak, amelyek legalább egy nitrogénatomot tartalmaznak. Ezek a vegyületek összefoglaló néven az azolok. Elméletileg a furán vagy tiofén vagy pirrol egy vagy több „CH”-csoportját nitrogénatommal helyettesítve jutunk az azolok gyűrűvázához. A belépő nitrogén bázisos jellegű, mivel elektronpárja nem vesz részt a π-elektronrendszer kialakításában.
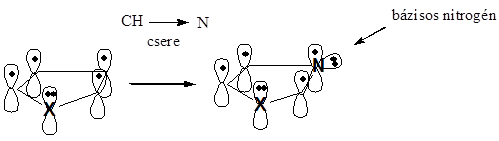
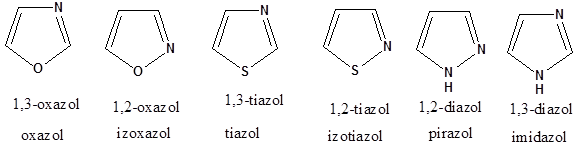
Az azolok különböző karbonsavszármazékokból és 2-klórketonból nyerhetők:
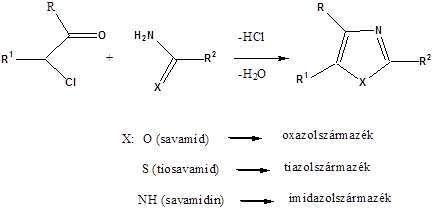
A pirazolszármazékok jó hozammal nyerhetők 1,3-dikarbonilvegyületekből hidrazinnal, illetve alkil- és arilhidrazinokkal végzett ciklokondenzációs reakcióval:
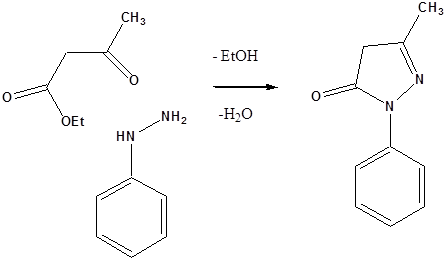
A pirrolból levezethető pirazol és imidazol, a „pirrol”-szerű savas N-H miatt amfoter. Ennek a két vegyületnek a forráspontja lényegesen magasabb, mint az N-H-t nem tartalmazó egyéb azoloké. Az ok: lineáris vagy gyűrűs N-H---N típusú hidrogénhid.
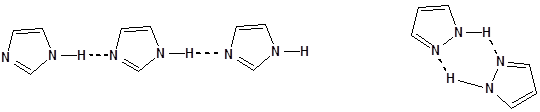
A nitrogénatomok közötti gyors protoncsere a nitrogénatomokat, s így a két ún. annuláris tautomert megkülönböztethetetlenné teszi (virtuális automéria). A 4- vagy 5-helyzetben monoszubsztituált imidazoloknak két valódi tautomer formájuk létezik, a 3- vagy 5-helyzetben szubsztituált pirazoloknak úgyszintén. A gyors protoncsere miatt ezeket a tautomereket egymástól általában nem lehet elválasztani. Ezt a tényt a vegyületek nevében is feltüntetjük: pl. 4(5)-metilimidazol.
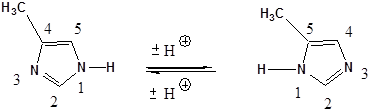
Aromás elektrofil szubsztitúcióban a reakciókészség valahol a piridin (ld. később) és az öttagú egy heteroatomos rendszerek között foglal helyet. Savas közegben az azolok bázikus nitrogénje protonálódik, a keletkező heterociklusos kation erősen dezaktivált (pl. nitrálósavval az imidazol 9 nagyságrenddel lassabban nitrálódik mint a benzol), ezért savas közegben végbemenő szubsztitúciókat csak erélyes körülmények között lehet végrehajtani. Semleges közegben a reakcióképességük közel összemérhető a benzoléval. A savas NH-csoportot tartalmazó pirazolból és imidazolból bázikus közegben képződő anion reakcióképessége ellenben jelentősen nagyobb (9-12 nagyságrenddel), mint a semleges heterociklusé.
Az 1,2-azolok reakciókészségi sorrendje: pirazol > izotiazol > izoxazol. A szubsztitució elsősorban a 4-es helyzetben játszódik le.
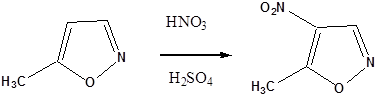
Az 1,3-azolok SEAr reakciókészsége, mely nagyobb, mint a megfelelő 1,2-azoloké, az imidazol > tiazol > oxazol sorrendben csökken.
Az oxazol és a tiazol az 5-helyzetben szubsztituálódik. Savas és semleges közegben az imidazol elektrofilekkel 4(5)-helyzetben reagál. Lúgos közegben, a képződő konjugált bázis legreakcióképesebb helye a 2-es.
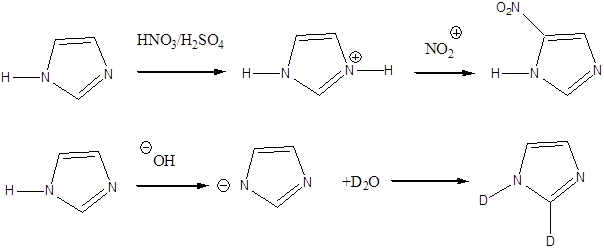
A hisztidin 3-[imidazol-4(5)-il]alanin (-)-(S)-enantiomer. Természetes aminosav. Dekarboxilezése hisztaminhoz vezet, melynek mennyisége, ha a szervezetben túlzottan megnő, allergiás tüneteket okoz. Az 5,5-difenilhidantoint régebben antiepileptikumként használták.
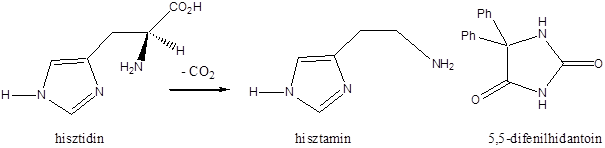
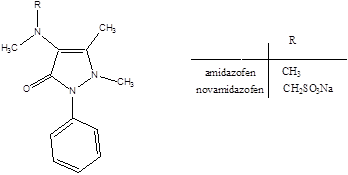
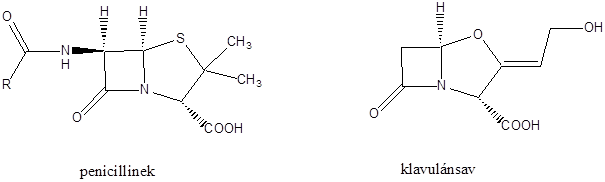
Tartalom
A hattagú, egy oxigént tartalmazó maximális számú nem kumulált kettős kötést tartalmazó heterociklusos vegyület a pirán, amely nem aromás. A 2H- ill. 4H-piránból hidridion elvételével levezethető kation, a piriliumion a piridinnel izoelektromos, aromás vegyület.
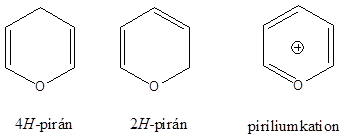
Flavonok, izoflavonok és favonolok. A pirán benzollal kondenzált származékai (benzopirán gyűrűrendszer). A természetben ezeknek az alapvegyületeknek hidroxi- és alkoxi-származékai fordulnak elő glikozidok formájában.
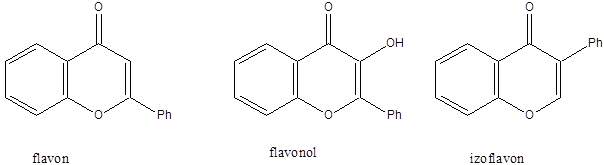
Antocianidinek és katechinek. A flavonokkal szerkezeti rokonságban és genetikus kapcsolatban álló anyagok. Az anticianidinek glikozidjai (antociánok) a virágok vörös és kék színes anyagai, a katechinek cserző anyagok.
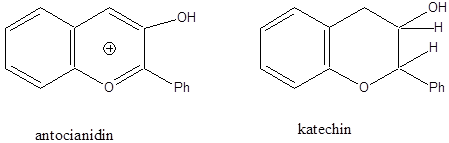
A természetben ezeknek a vegyületeknek a hidroxi-származékai fordulnak elő. Az antocianidinek színe pH függő. Savakban vörösek, lúgokban kékek.
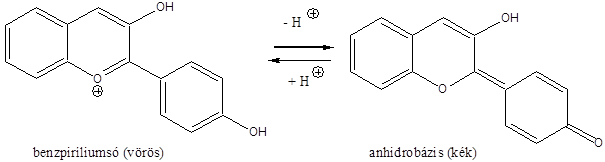
A benzol egy szénatomjának nitrogénre cserélésével levezethető heterociklus a piridin.
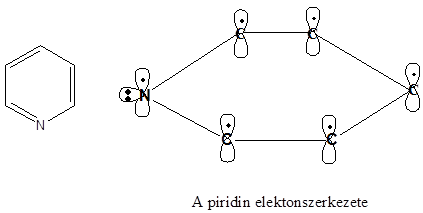
Mint a fenti ábrából kitűnik, a planáris σ-váz kialakulása után valamennyi gyűrűt alkotó atom rendelkezik erre a síkra merőleges pz pályával, s így létrejött 4n+2=6 db pz elektron részvételével a delokalizált aromás molekulapálya-rendszer. A nitrogén magános elektronpárja a gyűrű síkjától kifelé mutató MO-n helyezkedik el. Ennek köszönhető, hogy a piridin gyengén bázikus (pKa=5,2) és az, hogy a piridinium-sók is aromásak. A piridin delokalizációs energiája 117 kJ/mol, kisebb mint a benzolé (150 kJ/mol), ezért a dihidropiridinek stabilabbak mint a megfelelő ciklohexadiénszármazékok.
A piridin oxidációnak ellenáll, SEAr reakcióban a benzolnál lényegesen nehezebben szubsztituálható. Ennek oka egyrészt az, hogy a szénnél elektronegatívabb nitrogén a gyűrűben lévő elektronsűrűséget csökkenti, különösen 2- és 4-helyzetben. Az aromás elektrofil szubsztituciók egy jelentős része erősen savas körülmények között játszódik le. A piridin protonálódik, s ezáltal a nitrogén elektronszívó hatása tovább fokozódik. A piridin aromás nukleofil szubsztituciós reakcióban aktívabb a benzolnál.
A piridinszármazékokat Hantzsch módszerével, aldehidekből és β-oxo-észterekből, vagy –nitrilekből, valamint ammóniából állítjuk elő. A keletkező 1,4-dihidropiridint pl. mangán- dioxiddal oxidálhatjuk.
Csak igen erélyes körülmények között mennek végbe a gyűrű 3-as helyzetében.
A piridin-N-oxidja aromás elektrofil szubsztitucióba lényegesen könnyebben vihető, és 2- ill. 4-helyzetben szubsztituált piridinek állíthatók elő.
Az N-oxidokból az oxigén több módszerrel is eltávolítható, és így a szubsztituciós reakció után a 2- ill. 4-szubsztituált piridinhez jutunk.
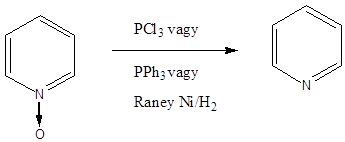
Az elektrofil szubsztitucióra dezaktivált szénatomokon a nukleofilek addíciója (így a szubsztitució is) viszonylag enyhe körülmények között végbemegy.
Csicsibabin-reakció: Addiciós-eliminációs mechanizmus szerint SNAr-reakcióra aktiváló szubsztituens jelenléte nélkül is lejátszódik.
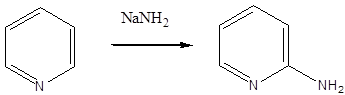
Piridinium-sók reakciói: A piridin N-alkilezésével előállítható piridínium-sók nukleofilek, addíciójára hajlamosak. A hidroxidion addíciójával keletkező 1-alkil-1,2-dihidropiridin-2-ol instabil vegyületet, savas közegben visszaalakul a kiindulási piridínium-sóvá (azaz pszeudobázis). Ha a hidroxidion nukleofil addícióját a Fe(III) só jelenlétében végezzük, az addíciót oxidáció követi.
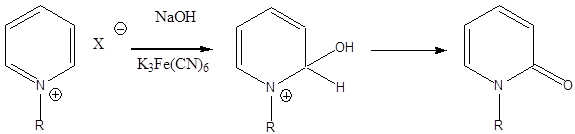
Piridin: jellemző szagú folyadék, Fp.: 115 oC, vízzel korlátlanul elegyedik, jó oldószere a szerves vegyületeknek és számos szervetlen sónak. Előfordulás: csontolaj, kőszénkátrány. Kinyerés: bázisos jellege folytán kivonás savval, majd elválasztás homológjaitól frakcionált desztillációval.
Nikotin a dohányzásban szerves savakkal alkotott sói formájában előforduló alkaloid. A tiszta nikotin balraforgató, csípős ízű, színtelen, levegőn megbarnuló folyadék. Idegméreg. Dohányosoknál a szervezet alkalmazkodása miatt a központi idegrendszerre ható, stimuláló hatású anyag. Felhasználás: inszekticid (rovarölő) hatása miatt növényvédő szer.
INH (izonikotinsav-hidrazid). Fontos antituberkulotikum.
B6-vitamin (5-hidroxi-6-metilpiridin-3,4-dimetanol) a transzaminázok prosztetikus csoportja.
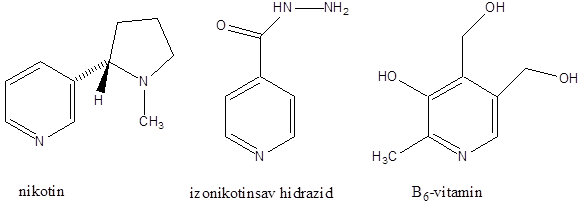
A piridin benzollal kondenzált származékai a kinolin és az izokinolin.
Kinolinvázas alkaloidok közé tartozik a kinin és rokon alkaloidok. A kininnek maláriaellenes hatása van. Az izokinolinvázas alkaloidok közé tartoznak a mák-alkaloidok: morfin, kodein, narkotin, papaverin. A papaverin simaizom görcsoldó.
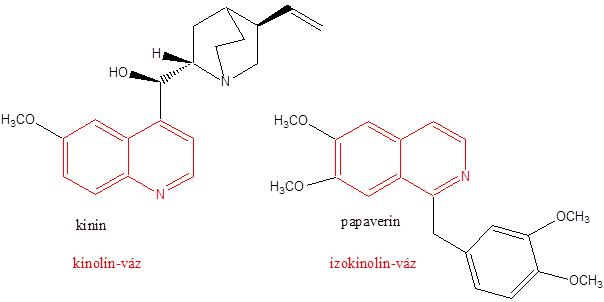
A hattagú, több heteroatomot tartalmazó heteroaromás vegyületek legfontosabb képviselői diazinok. Fenti képleteik alapján, a két nitrogénatom egymáshoz viszonyított helyzete szerint három alaptípusuk van, mégpedig triviális nevükön: piridazin, pirimidin és pirazin.
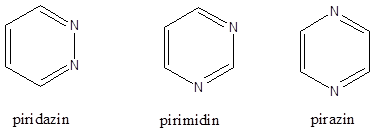
Piridazinszármazékok: 1,4-dioxovegyületekből hidrazinhidráttal könnyen nyerhetők. Először a megfelelő szubsztitúciójú dihidropiridazin-származék képződik, melyet enyhe oxidálószerekkel könnyen lehet piridazinszármazékokká alakítani. Az 1,2-dioxovegyületek és etiléndiaminok reakciója pirazinokat eredményez.
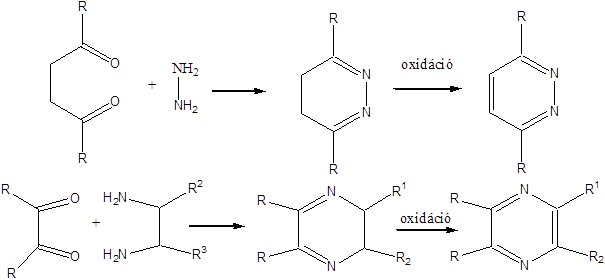
Pirimidinek: előállításukra számos módszer ismeretes. Többnyire a reakciópartnerek egyike tartalmazza mindkét nitrogénatomot, a másik komponens egy 1,3-bifunkciós elektrofil. Ezen az úton – megfelelő komponensek megválasztásával – a pirimidingyűrű különböző pozícióban különféle helyettesítőket tartalmazó származékok, így például pirimidinbázisok, barbitursav származékok állíthatók elő.
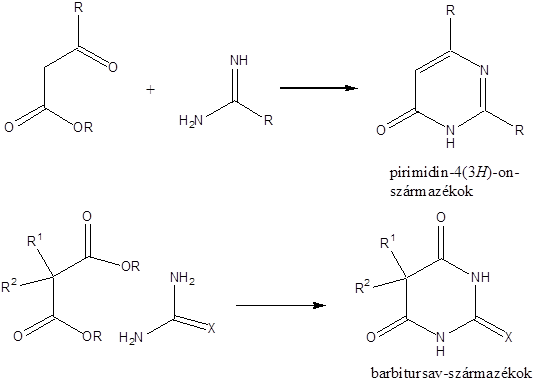
A diazinok elektrofilekkel SEAr reakcióban a piridinnél jóval kisebb reakciókészséget mutatnak. A pirimidingyűrű dezaktivált, ugyanakkor egy vagy két aktiváló szubsztituens (pl. amino- és/vagy hidroxilcsoport) a 2-, 4- vagy 6-helyzetben már lehetővé teszi a nitrálás, nitrozálás, aminometilezés, halogénezés és azokapcsolás lejátszódását. Megjegyezzük azonban, hogy ezek a reakciók rendszerint a szubsztituált pirimidingyűrű tautomer formáin, addíciós –eliminációs mechanizmus szerint mennek végbe.
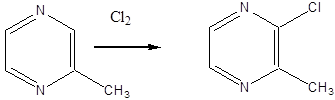
A hattagú, két heteroatomos nitrogéntartalmú vegyületek bázikus tulajdonságúak. Bázicitásuk mértékét a konjugált sav aciditásával (pKa) jellemezzük. A bázicitás a két nitrogénatom egymáshoz viszonyított jellegétől függ. Az 1,2-helyzetben lévő két nitrogénatom nemkötő elektronpárjai a térközelség miatt jelentősen taszítják egymást. Ezért, ha az egyik nitrogénatom protonálódik, az elektronpárok taszítása jelentősen csökken, ami energetikai szempontból előnyös. Ez a hatás fokozatosan csökken a két nitrogénatom távolságának növekedésével, mivel a protonálódás következtében fellépő energianyereség egyre kisebb. Ezért az 1,2>1,3>1,4 diazin sorrendben csökken a vegyületek bázicitása, azaz így növekszik a konjugált sav aciditása, pKa = piridazin: 2,33; pirimidin: 1,30; pirazin: 0,65.
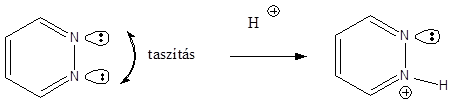
A diazinok nukleofil ágensekkel szemben mutatott fokozott reakciókészsége széles körben kiaknázható. Szintetikus szempontból fontosak a diazinok halogénszármazékainak SNAr reakciói, mégpedig a halogénszármazékok jó hozzáférhetősége és a halogént viselő szénatom fokozott elektrofilitása miatt. Különféle halodiazinok 4-nitrofenoxiddal szemben tapasztalt reaktivitási sorrendjéből kitűnik, hogy általában a gyűrűnitrogénekhez orto- és/vagy para-helyzetben lévő halogének a legreaktívabbak a pirrimidin > piridazin > pirazin sorrendben.
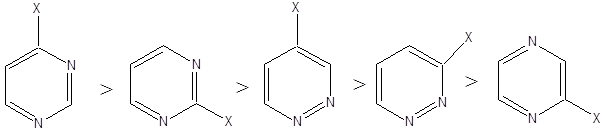
A három diazin közül a pirimidin származékai a legelterjettebbek. A DNS és RNS felépítésében kulcsfontosságú szerepük van. A DNS-ben és az RNS-ben is a pirimidingyűrű aminocsoportokat és hidroxilcsoportokat tartalmaz. Ezeknél a vegyületeknél laktim/laktám, ill. imino/amino tautomériával kell számolni. Modellvegyületnek tekintsük a 2-hidroxipiridint, amelynek két tautomerjét és egy ikerionos (rezonancia) formáját rajzolhatjuk fel:
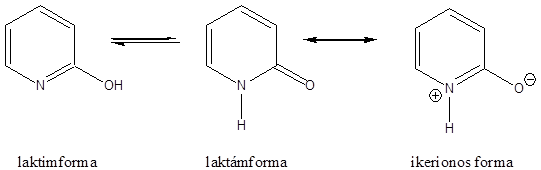
A spektroszkópiai vizsgálatok azt mutatták ki, hogy a 2-hidroxipiridin és 4-hidroxipiridin kristályos állapotban, valamint kloroformos oldatban csaknem teljes mértékben piridonszerkezetű. Ezzel szemben mind lúgos oldatban, mind pedig gázfázisban a hidroxipiridin-szerkezet az uralkodó.
Két heteroatomot tartalmazó vegyületek. A pirimidinszármazékok esetében a 2-helyzetben szubsztituált vegyület tautomer formáit mutatjuk be. Hasonlóan a piridin-2(1H)-on származékához, poláris közegben az oxoforma, apoláris körülmények között a hidroxiforma van jelen.
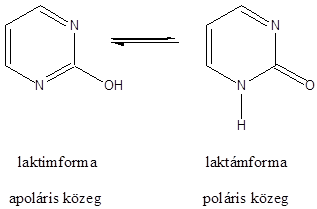
A másik két diazin analóg szerkezetű oxoszármazékai [piridazin-3(2H)-on és a pirazin-2(1H)-on] esetében is hasonlóan alakul a tautomer egyensúly helyzete. Az aminocsoport viszont általában nem tautomerizálódik iminoformává.
Pirimidingyűrűt tartalmaz számos élettani szempontból fontos más anyag is, így például a B1-vitamin, mégpedig pirimidin- és tiazol-gyűrűt.
Ugyancsak élettani jelentőségű a diazino-diazinok közé tartozó pteridin, és származékai a folsav és a methotrexat.
A folsav részt vesz a nukleotid bioszintézisben, továbbá biológiai redoxrendszerekben is szerepel. A methotrexat a folsavantagonisták csoportjába tartozó citosztatikum (sejtosztódást gátló, rákellenes szer).
A purinvázas vegyületek a nukleinsavakban fordulnak elő de más természetes forrásaik is vannak. A purinnak (szisztematikus neve: imidazol[4,5-d]pirimidin) két tautomerje létezik, a 7H-purin és a 9H-purin-forma.

A legrégebben ismert purinvázas vegyület a húgysav (2,6,8-trihidroxipurin).
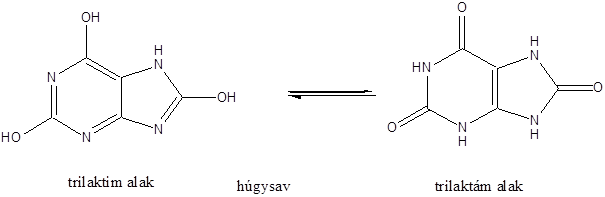
További purinvázas vegyületek a a DNS-ben ill. RNS-ben megtalálható adenin és guanin (ld. Nukleinsavak). A purinvázas alkaloidok a 7H-purin származékai. A teofillin a teacserjében, a teobromin a kóla és kakaó növényekben, a koffein a kávéban és teacserjében fordul elő.
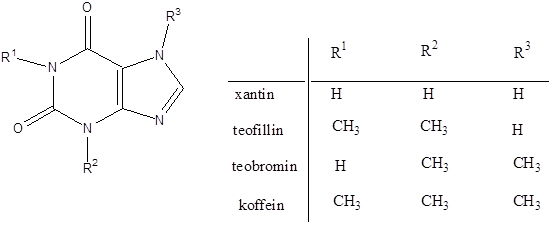
A citosztatikumokként alkalmazható ún. antimetabolitok körében ugyancsak vannak purin-, valamint pirimidinvázas vegyületek. Az előbbiekre a 6-szulfanilpurin (merkaptopurin), utóbbiakra az 5-fluorouracil (fluorouracil) a példa. Az aciklovir antivirális szer, amit herpesz és övsömör kezelésére használnak.
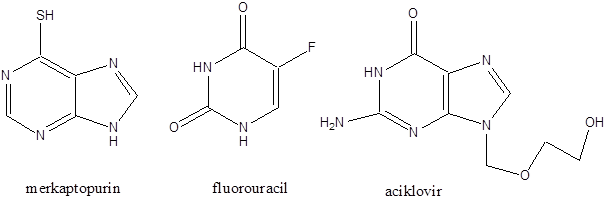
Tartalom
A nukleotidok szerepe a biokémiai folyamatokban igen széleskörű:
- a nukeinsavak (DNS, RNS) alapegységei
- energiaforrások (ATP, GTP)
- regulációs molekulák (cAMP)
- koenzim komponensek (NAD, FAD, koenzim-A)
- származékaik a szénhidrát- és lipidanyagcsere intermedierjei (UDP-galaktóz, CDP-diacil-glicerin)
Minden nukleotid három alkotóból épül fel: egy heterociklusos bázisból (purin vagy pirimidin), egy 5 szénatomos cukorból (ribóz vagy dezoxiribóz) és egy foszforsavból.
A bázisrész pirimidin- vagy purinszármazék. A pirimidinbázisok közül legfontosabbak az uracil, a citozin és a timin (metiluracil). A legfontosabb purinbázisok az adenin és a guanin. Mind a purin-, mind a pirimidinbázisok laktám-laktim tautomer formában fordulnak elő, de fiziológiás körülmények között a laktám forma dominál, ami a pirimidinek esetén az N-glikozidos kötés kialakulásának az előfeltétele.
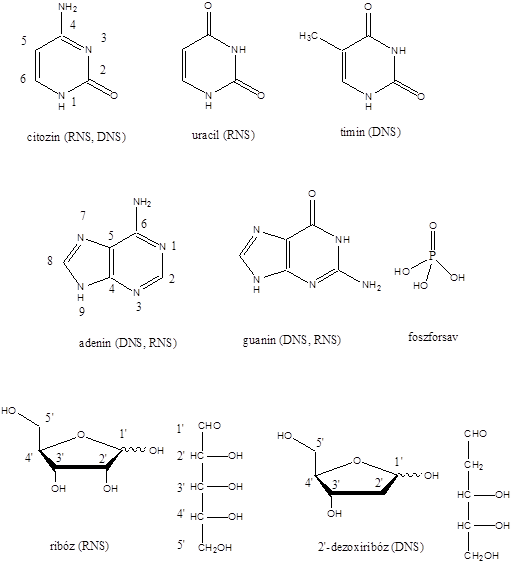
A bázisok -N-glikozidos kötéssel a pentózok (ribóz vagy dezoxiribóz) C1-atomjához (1’C) kapcsolódva alakítják ki a nukleozidokat A cukorrésztől függően ribonukleozidok vagy dezoxiribonukleozidok jöhetnek létre. A foszforsav a cukormolekula C5-atomján (5’C) lévő OH-csoportot észteresíti. A nukleozidok 5’-foszfátésztereit nukleotidoknak nevezzük nukleozidok és nukleotidok elnevezéseit és jelöléseit mutatja a következő ábra:
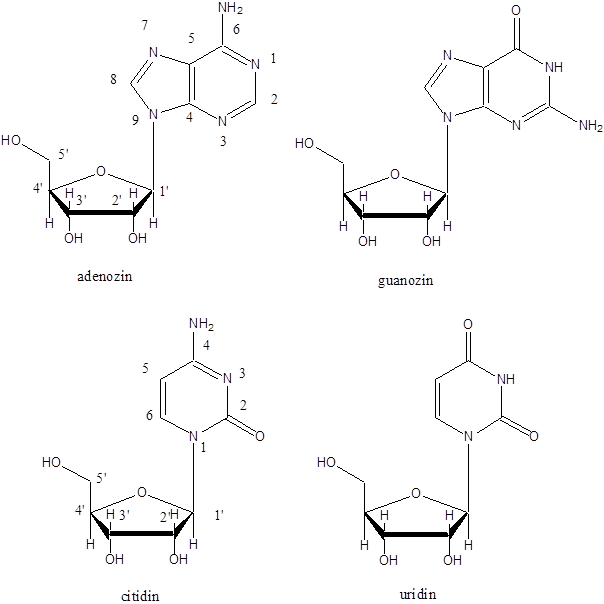
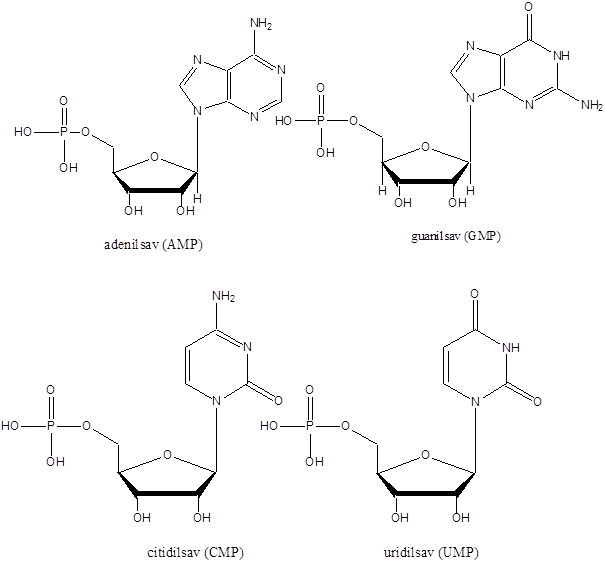
Az 5’-nukleozid-monofoszfátok savként viselkednek pKa értékei 1 és 6 körüliek, azaz vizes közegben dianionként vannak jelen. Az 5’-nukleozid-monofoszfát foszfátcsoportjához nagy energiájú savanhidrid-kötéssel további foszfátcsoportok is kapcsolódhatnak és létrejöhetnek a di- és trifoszfátok (pl. ADP, ATP) amelyek az élő szervezetek energiatároló molekulái.
A monofoszfátokból létrejöhet ciklikus szerkezetű vegyület is, ha egy foszfátcsoportot a ribóz 3’-és 5’-OH-ját összekötve észteresíti. Az egyik legfontosabb képviselőjüket a ciklikus AMP (cAMP), aminek a biológiai folyamatok szabályozásában van nagy szerepe.
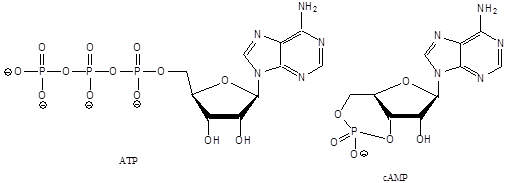
A dinukleotidok igen fontos szerepet játszanak a az élőszervezetek redox folyamataiban, a nikotinamid adenin dinukleotid (NAD+) a dehidrogenázok koenzimje, képes proton és két elektron felvételével NADH-vá alakulni.
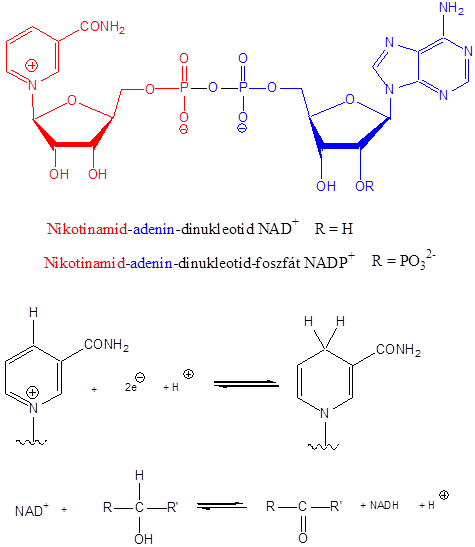
A flavin-dinukleotid szintén redox folyamatokban vesz részt, pl. a borostyánkősavat fumársavvá dehidrogénezi.
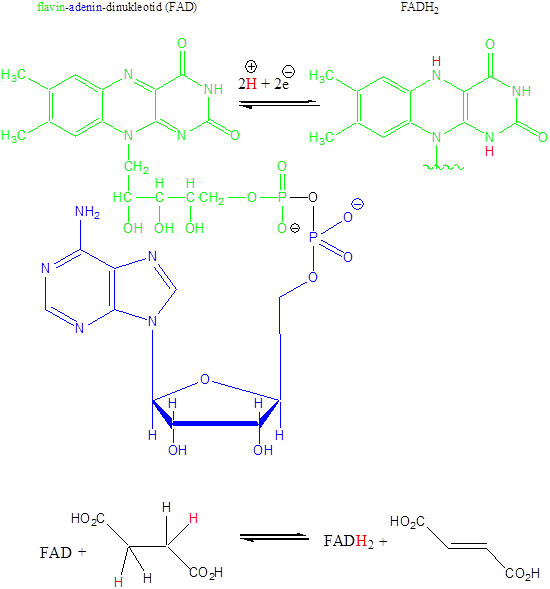
A nukleinsavak nukleotid egységekből álló lineáris, el nem ágazó polimerek. A ribózt tartalmazó polinukleotid a ribonukleinsav (RNS), a dezoxiribóz tartalmú a dezoxiribonukleinsav (DNS). A DNS tárolja az élőlények genetikai információit. A különböző RNS-ek ezen információk kifejeződésében, elsősorban fehérjeszintézisben játszanak fontos szerepet.
Miescher már 1869-ben felfedezte a nukleinsavat, de kémiai felépítését csak az 1950-es években tudták meghatározni. A DNS-ben előforduló purin bázisok az adenin (A) és a guanin (G), a pirimidin bázisok a citozin (C) és a timin (T), a cukorrész a dezoxiribóz. Az RNS-t felépítő bázisok az A, G, C és az uracil (U), míg a cukorrész a ribóz. A két nukleinsav között kémiai összetételüket illetően kicsi az eltérés, ugyanakkor a biológiai szerepük jelentősen eltér egymástól.
A nukleinsavak (DNS, RNS) elsődleges szerkezetén a nukleotidsorrendet értjük. A polinukleotid lánc ún. 5’-végét képező nukleotid cukorkomponensének 5’-OH-ja többnyire foszfáttal észteresített formában van, míg 3’-végét alkotó nukleotid cukorkomponensének 3’-OH-ja többnyire szabad. Az általánosan elfogadott konvenció alapján a polinukleotidok bázisainak sorrendjét az 5’-végtől a 3’-vég felé írjuk fel. Általában az egyszerűsített írásmódnak megfelelően a bázisokat betűvel, a cukorrészt függőleges vonallal, az egységeket összekötő foszfodiészter-kötést ferde vonal közötti P betűvel jelöljük.
Az igen hosszú DNS-ek méretének jellemzésére megállapodás szerint a bázispárok (bp) számát, vagy ennek ezerszeresét, a kilobázispárok (kb) számát adjuk meg. Például az E. coli DNS mérete 4600 kb, az élesztőé 12600 kb (16 haploid kromoszómában ) és az emberi DNS hossza 2900000 kb (23 haploid kromoszómában). (Haploid = egyszeres, a szokásos számának fele).
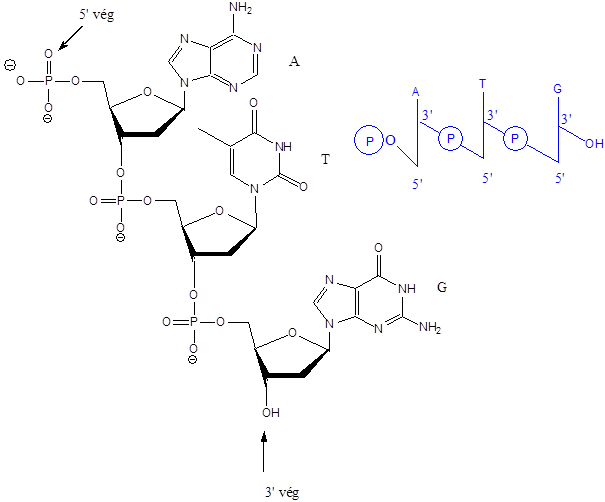
A DNS másodlagos szerkezetét a két ellentétes irányultságú polinukleotidlánc közös tengely köré csavarodó kettős spirálja (hélix) adja. A DNS kettős hélix szerkezetének felderítése James Watson és Francis Crick nevéhez fűződik. Munkájuk eredményességét számos kutató segítette, például Erwin Chargaff (Chargaff-szabályok), aki többek között megállapította, hogy minden esetben az adenin mennyisége egyenlő a timinével (A=T), illetve a guanin mennyisége egyenlő a citozinéval (G=C). Az A és T valamint a G és C aránya 1:1. Jerry Donohue (1920-1985) munkája bizonyította, hogy a bázisok laktám-tautomer formában léteznek, ami feltétele a bázisok közötti hidrogénhidak kialakulásának. Rosalind Franklin röntgendiffrakciós vizsgálatai alapján derült fény a DNS helikális szerkezetére.
Mindez alapján 1953-ban Watson és Circk az ún. B-konformációjú (jobbmenetes) DNS szerkezetét írta le. A kettős hélix átmérője 2 nm, egy menet magassága 3,4 nm és kb. 10 bázispárból áll, a bázisok síkja egymástól 0,34 nm távolságra helyezkedik el. A két polinukleotidban a mononukleotidok 3’,5’-foszfodiészter-kötéssel kapcsolódnak egymáshoz úgy, hogy az egyik nukleotid dezoxiribózának 3’-hidroxilcsoportjához egy másik nukleotid cukoregységének 5’-hidroxilcsoportja egy foszfátcsoporton keresztül kapcsolódik. A dezoxiribóz-foszfodiészter kötések alkotják a kettős hélix vázát, a bázisok pedig a hélix belseje felé nyúlnak. A bázispárokat hidrogénkötések tartják össze, amelyek az adenin és timin (két hidrogénhíd), valamint a citozin és guanin (három hidrogénhíd) között létesülnek A kettős hélix külső részét képező cukor-foszfát-váz csavarulatai között két helikális árok helyezkedik el, a szélesebbet nagy ároknak (2.2 nm), a keskenyebbet kis ároknak (1,2 nm) nevezzük.
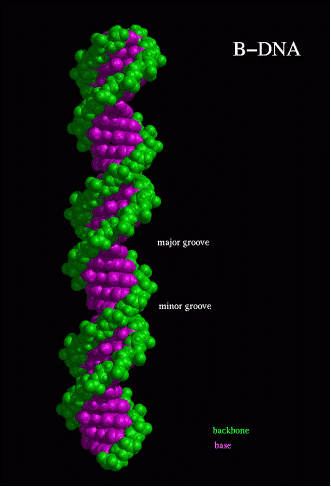
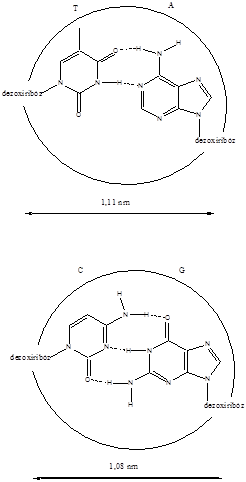
A DNS számos konformációt vehet fel a nukleotid szekvencia függvényében A B-DNS mellett A-DNS, C-DNS és Z-DNS formák is léteznek. A B-formában 10 bázispár alkot egy csavarulatot. A bázisok a hélix tengelyhez közel helyezkednek el, de arra nem teljesen merőlegesen, az eltérés kb. 6o. Az A-formában 11 nukleotidpár alkot egy csavarulatot. A bázisok a hélix tengelyétől távolabb helyezkednek el és a tengely felé hajlanak, így a hélix megrövidül. A C-formánál egy csavarulatban 9 bázispár van. A Z-forma onnan kapta a nevét, hogy a szerkezet cikk-cakk alakú és a hélix balmenetes. A Z-formában 12 nukleotidpár alkot egy csavarulatot, a DNS hosszabb és vékonyabb, mint a B-forma.
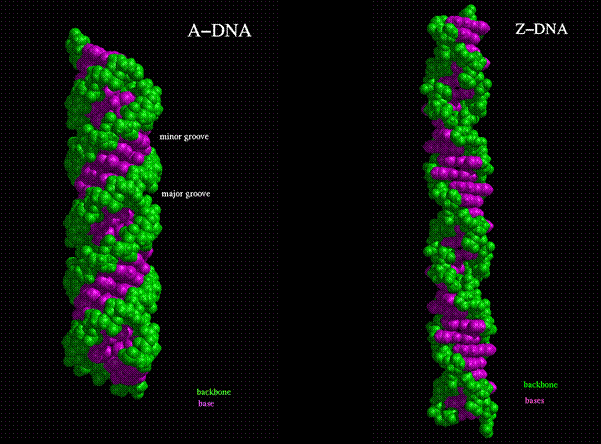
A DNS harmadlagos szerkezetének az a felcsavarodott szuperhelikális forma tekinthető, amely a magnélküli (prokarióta) sejtekben a cirkuláris és a sejtmaggal rendelkező (eukarióta) sejtekben lineáris formájú DNS-ekből jön létre annak érdekében, hogy a DNS elférjen a sejtben. Az eukarióta DNS fehérjékkel komplexeket képezve és többszörösen feltekeredve alkotja a kromatinállományt, amit a DNS negyedleges szerkezetének nevezhetünk. A kromatinban a hiszton gyöngysor egy-egy szeme négyféle hisztonból (H2A, H2B, H3, H4) áll, ami a hozzátartozó DNS darabbal együtt alkotja a nukleoszómát. Egy nukleoszómában a 4-féle hisztonmolekulából kettő-kettő fordul elő, és a DNS kétszer tekeredik a nyolc alegységes mag köré. A nukleoszómasor felületén tekeredő DNS-szálhoz kívülről egy ötödik hiszton (H1) kötődik, stabilizálva a kromatint.

Az RNS elsődleges szerkezetét, a DNS-hez hasonlóan, a nukleotidsorrend határozza meg. A legtöbb RNS egyenes láncú ezért a bázisok aránya tetszőleges. A sejtekben többféle RNS is előfordul, ezek molekulatömege kisebb mint a DNS molekulatömege.
A riboszómális RNS-ek (rRNS) a sejt RNS-állományának mintegy 75%-át teszik ki. A riboszómákban találhatók, 120-5000 nukleotid egységből épülnek fel, molekulatömegük 40000-150000 Da (dalton) közé esik.
A transzfer RNS-ek (tRNS) 70-90 nukleotid egységből épülnek fel, ezek a legkisebb RNS-ek (Mt:23000-25000 Da). A sejtek RNS állományának 10-20%-át teszik ki. A tRNS-ek jellegzetes „lóhere alakú” másodlagos szerkezettel rendelkeznek, aminek révén kitüntetett szerepet játszanak a fehérje bioszintézisben. A tRNS-ek specifikusan kötik és szállítják az aminosavakat a riboszómához kötődő mRNS megfelelő bázishármasához. Minden aminosavat más-más tRNS szállít. A sejtben kb. 60-féle tRNS található.
A messenger vagy hírvivő RNS-ek (mRNS) a sejtek RNS-állományának kb. 5%-át teszik ki. Molekulatömegük 250000-1000000 Da közé esik. Az mRNS nukleotid szekvenciái tartalmazzák a templátot (mintát) a polipeptidláncok szintéziséhez. A prokarióták mRNS-e policisztronos, azaz több fehérje aminosav-sorrendjére vonatkozó információt hordoz, míg az eukariótáké csupán egy fehérje bioszintéziséhez szolgál alapul (monocisztronos).
A kis nukleáris RNS-ek (snRNS = small nuclear RNS) a transzkripció utáni módosításban vesznek részt, vagy specifikus fehérjékkel képeznek komplexeket (ribonukleoproteidek).
Az újabb felfedezéseknek köszönhetően egyre több RNS-funkcióra és RNS-formára derül fény, amelyek nem a fehérje bioszintéziséhez kötődnek. Például a miRNS (micro) az mRNS lebontásában, a siRNS (small interfering) a génműködés szabályozásában, a telomeráz RNS a replikációban vesz részt. Az RNS-ek változatos másodlagos szerkezetekkel rendelkeznek, egyedül a tRNS-nek van jellegzetes (lóhere alakú) szerkezete.
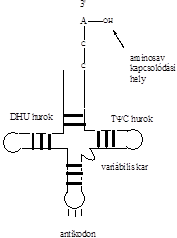
A molekuláris biológia „centrális dogmája” szerint a DNS-ben tárolt információ nem alkalmas arra, hogy a fehérjék szintéziséhez templátként szolgáljon, hanem szükség van a mRNS-re is. Crick 1958-ban megállapította az információ áramlás irányát, amely DNSfehérje. A fehérje nem lehet a genetikai információ tárolója, azaz DNS vagy RNS templátja. Egyedül bizonyos vírusok képesek arra, hogy RNS-ből DNS-re átírjanak információt (reverz transzkripció).
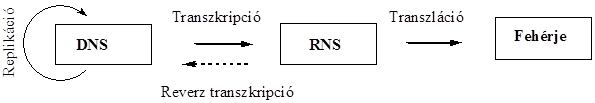
Nevezze el az alábbi vegyületeket:
2.) Az enteroszeptor fertőtlenítő szer neve: 8-hidroxi-7-jód-5-klórkinolin. Írja fel a vegyület szerkezeti képletét.
3.) Mi lesz az alábbi reakciók terméke? Írja rá a szaggatott vonalakra!
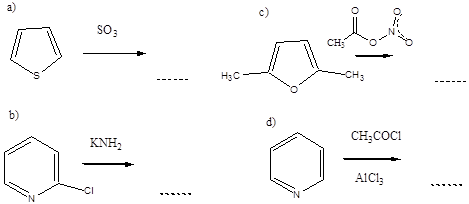
4.)Az alábbi tautomer párok közül válassza ki a poláros oldószerben stabilisabbat:
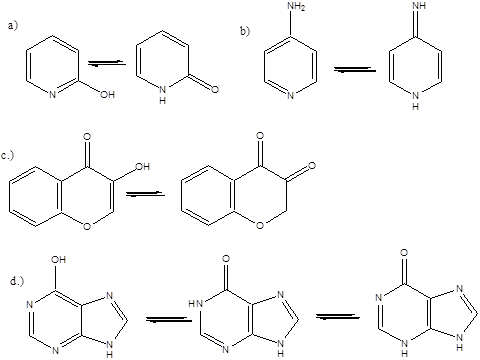
5.) Állítsa elő a következő vegyületeket Fischer-féle indolszintézissel:
a.) 2-metilindol b) 2-fenilindol
6.) Hogyan állítana elő 2,6-dietil-4-fenilpiridint Hantzsch-féle piridinszintézissel?
7.) Szintetizálja a pentobarbital (Nembutal) gyógyszert karbamidból és dietil-malonátból kiindulva!
8.) A piridint lúgos kálium-permanganát oldattal melegítve nem történik változás. Ha az a kísérletet savas kálium-permanganáttal ismételjük meg barna csapadék jelenik meg! Értelmezze a tapasztalatot!
9.) A 3-etinilpiridint nátrium-amiddal hevítve egy C7H6N2 összetételű vegyületet kapunk, ami nem tartalmaz CC kötést. Mi lehet a vegyület szerkezete? A keletkezett vegyületben melyik nitrogén bázisosabb?
10.) Az 5’-AATGCCGACT-3’ DNS szálnak írja fel a komplementer DNS és a komplementer mRNS megfelelőjét!
11.) Milyen hexapeptidet kódol az alábbi mRNS szakasz? (Használjon genetikai kódszótárt!)
CUA-GAC-CGU-UCC-AAG-UGA
12.) A citozin salétromossav hatására uracillá alakul. Miért és hogyan játszódik le a reakció? Mi a citozin bázispárja? Mi lesz az uracil bázispárja?
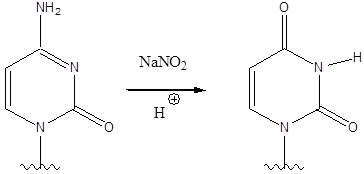
13.) Az AIDS megbetegedés kezelésére lassítására a 3’-azidotimidint használták (AZT). Rajzolja fel a szerkezetét! Min alapszik az AZT hatása?
14.) A tengeri sün DNS-e 32% adedint tartalmaz? Mi lesz a másik 3 bázispár %-os aránya?
Tartalom
A vitaminokat többféle szempont szerint csoportosíthatjuk:
- oldhatóság
- biokémiai szerep
- kémiai szerkezet stb.
A legáltalánosabban elfogadott az oldhatóság szerinti csoportosítás:
1. zsíroldható vitaminok
2. vízoldható vitaminok
Az ismert vitaminok a következők:
-
zsírban oldható vitaminok
A-vitaminok (antixeroftalmiás vitaminok)
D-vitaminok (antirachitiszes vitaminok)
E-vitaminok (szaporodási vitaminok)
K-vitaminok (antihemorrágiás vitaminok).
-
Vízben oldható vitaminok:
B1-vitamin (antineuritiszes vitamin), tiamin
B2-vitamin, riboflavin
B3-vitamin (antidermatitiszes tényező), pantoténsav
B5-vitamin (antipellagrás vitamin) nikotinsavamid
B6-vitamin (antidermatitiszes vitamin), piridoxin
B7-vitamin (antidermatitiszes tényező), biotin
B10-vitamin (antianémiás vitamin), folsav
B12-vitamin, (antianémiás vitamin), cianokobalaminok
C-vitamin (antiskorbutos vitamin), aszkorbinsav
U-vitamin (antiulcus tényező), S-metil-metionin
|
Betűjel, név és képlet |
Természetes forrás |
Hiánybetegség |
Közepes napi szükséglet (mg) |
|
A, retinol
|
Tej, tojás, máj, gyümölcsök, zöldségek, halak májolaja |
xeroftalmia szemszárazság hiperkeratózis (szaruréteg túlburjánzás) |
1,5-2 |
|
D2, (ergokalciferol)
|
Hal, csukamájolaj, napfény hatására is képződik |
rachitis (angolkór) |
0,025 |
|
E, tokoferol
|
Búzacsíraolaj magvak |
szaporodási zavarok |
(20) |
|
K1, fillokinon
|
Szójaolaj, spenót, káposzta |
véralvadási zavarok |
(0,1) |
|
B1, tiamin
|
Korpa, hús |
beri-beri polineuritis (ideggyulladás) |
1-2 |
|
Betűjel, név és képlet |
Természetes forrás |
Hiánybetegség |
Közepes napi szükséglet (mg) |
|
B2 , riboflavin
|
tej, élesztő, máj,vese riboflavin, laktoflavin |
keratitis (szaruhártya-gyulladás), dermatitis (bőrgyulladás) |
1,5-2 |
|
B3, nikotinsavamid
|
Hering, élesztő, rizskorpa, szójabab, paraj |
pellagra |
15-20 |
|
B5, pantoténsav
|
Máj, tojássárgája, zöld növényi részek |
Burning-Feet- szindróma |
(10) |
|
B6, piridoxin
|
Hús, hal, élesztő |
epilepsziaszerű görcs, idegesség, álmatlanság,vérszegénység |
1-2 |
|
B7, biotin
|
Gyümölcs, főzelék, tojássárgája, máj, vese |
Dermatitisz, hámlás, szörzet hullása |
- |
|
B10 , folsav
n= 1, 3, 7 |
Zöld levelek, élesztő, vese, máj |
megaloblasztos anémia (vészes vérszegénység) |
(1-2) |
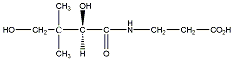
|
Betűjel, név és képlet |
Természetes forrás |
Hiánybetegség |
Közepes napi szükséglet (mg) |
|
B12, kobalamin
|
Máj, halliszt, tojássárgája |
anaemia perniciosa (vészes vérszegénység) |
(0,001) |
|
C, aszkorbinsav
|
Paprika, fekete ribiszke, csipkebogyó, savanyú káposzta |
skorbut |
75 |
|
U, S-metil-metionin
|
Növényi levelek, gyümölcslé |
emésztőrendszer fekélyei |
- |
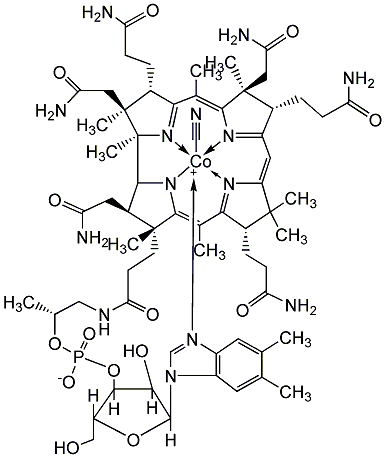
Tartalom
A kémiai ipar számos, a konvencionálisnál (pl. háztartási és mezőgazdasági hulladék) súlyosabb szennyeződést okozhat a környezetének, mind az üzemszerű működés folytán (pl. égéstermékek, melléktermékek kibocsátása), mind katasztrófák következményeként (pl. vörösiszap katasztrófa, a Tisza cianid szennyezése, tankerek katasztrófái stb.).
Ugyanakkor a kémia tudomány alkalmas arra, hogy e káros behatásokat mérsékelje vagy megszüntesse. Mindenesetre törekednünk kell arra, hogy a vegyipar minél környezetbarátabb folyamatokra térjen át, vagyis a zöld kémia (green chemistry) alábbi 12 pontja egyre nagyobb jelentőséggel bír.
1. Jobb megelőzni a hulladék keletkezését, mint azt keletkezése után kezelni.
2. Szintézisek tervezésénél törekedni kell a kiindulási anyagok maximális felhasználására, vagyis a nagyobb atomhatékonyságra.
3. Lehetőség szerint már a szintézisek tervezésénél olyan reakciókat célszerű választani, melyekben az alkalmazott és a keletkező anyagok nem mérgező hatásúak és a természetes környezetre nem ártalmasak.
4. Kémiai termékek tervezésénél törekedni kell arra, hogy a termékekkel szembeni elvárások teljesítése mellett mérgező hatásuk minél kisebb mértékű legyen.
5. Minimalizálni kell a segédanyagok használatát, ezek lehetőleg ne legyenek ártalmasak a környezetre.
6. Törekedni kell az energiafelhasználás csökkentésére.
7. A vegyipari alapanyagokat válasszuk megújuló nyersanyagokból.
8. Kerülni kell a felesleges származékkészítést.
9. Reagensek helyett szelektív katalizátorok alkalmazását kell előtérbe helyezni.
10. A kémiai termékeket úgy kell megtervezni, hogy használatuk végeztével ne maradjanak a környezetben és bomlásuk a környezetre ártalmatlan termékek képződéséhez vezessen.
11. Új és érzékeny analitikai módszereket kell használni a vegyipari folyamatok helyszíni ellenőrzésére, hogy a veszélyes anyagok keletkezését idejében észleljük.
12. A vegyipari folyamatokban olyan anyagokat kell használni, amelyek csökkentik a vegyipari balesetek valószínűségét.
Az antropogén szennyezőanyagok lebomlása vagy éppen akkumulációja a környezeti kémia, hulladék gazdálkodás igen fontos kérdései. Ebben a fejezetben néhány, a természetbe került szerves vegyület lebomlását és sok esetben súlyos környezetszennyezését tekintjük át.
Földi körülmények között a leggyakoribb szennyező anyagok a következő reakciótípusokban alakulhatnak át:
- fotokémiai folyamatok, a napenergiában gazdag spektrumtartománya révén;
- redoxireakciók főként a triplett, illetve szingulett molekuláris és atomos oxigén, továbbá az ózon segítségével;
- különböző pH tartományokban lejátszódó hidrolízis reakciók;
- reakciók a biológiai rendszerekben, amelyek az energiatermelő légzési folyamatokhoz kapcsolódva exergonikusan és endergonikusan is végbemehetnek,
Megjegyezzük, hogy sok estben e folyamatok kombinációi valósulnak meg a lebontási folyamatokban. A gyors és teljes átalakuláshoz vezető folyamatok esetében a környezetszennyeződés veszélye kicsi, az antropogén anyagok tökéletlen átalakulása azonban a szennyező anyag feldúsulását eredményezheti.
A környezetben lejátszódó fotokémiai reakciókat a 250-700 nm tartományba eső ultraibolya, illetve látható fény indítja el. Ha a kérdéses anyag ebben a tartományban abszorbeál, akkor energiában gazdagabb, gerjesztett állapotba kerül, amelyből mind intermolekuláris, mind intramolekuláris átalakulás révén deaktiválódhat. Ezen túlmenően gyakran van lehetőség arra is, hogy fotokémiai úton létrejövő, gerjesztett troposzféra-alkotórészekkel (O3, 1O2, stb.) játszódjék le a reakció.
A freonokat, mint illékony, kémiailag inert vegyületeket széles körben alkalmazták hűtőgépek, klímák hűtöközegeként, hőhordozójaként valamint spray hajtógázként. A Freon 12 UV sugárzás hatására klóratomra és difluór-klórmetil gyökre bomlik a sztratoszférában és itt reagál az ózonnal. A keletkező hipoklorit gyök újabb ózonnal reagálva oxigént és klóratomot ad. A klóratom lesz a láncvivő részecske:
A 1,1,1-triklór-2,2-bisz(4-klórfenil)etánt (DDT) ultraibolya fénnyel besugározva a vegyület gyorsan elbomlik. A bomlástermékek között 1,1-diklór-2,2-bisz(4-klórfenil)etánt (DDD), 1,1-diklór-2,2-bisz(4-klórfenil)etént (DDE) és ketonokat azonosíthatunk. Ezek alapján feltételezhetjük, hogy első lépésben a C-Cl kötés homolitikus hasadása következik be, majd a képződött gyökök hidrogénlehasítás, illetve elektron átvitel közben sósavat, DDT-t és DDE-t adnak.
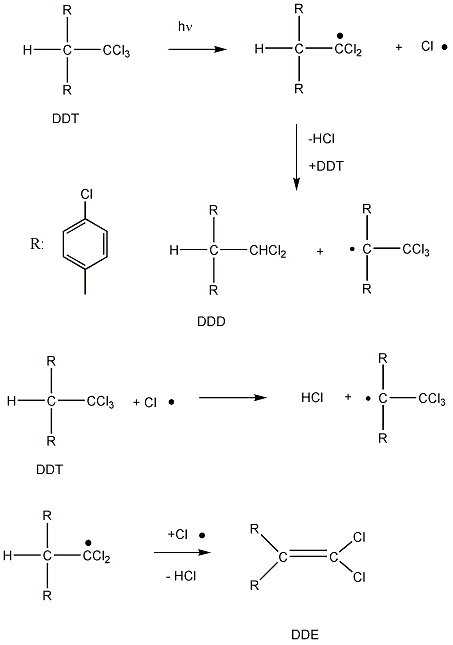
Ezek a termékek ugyanazok, mint amelyek a DDT mikrobiológiai lebontása során is keletkeznek. A malária és a sárgaláz kórokozója ellen kiváló rovarölő szernek bizonyult a DDT, azonban hosszabb ideig tartó használata alatt a melegvérűek zsírszöveteiben kimutathatóan felhalmozódott. A DDE gátolja pl. a szénsavanhidráz enzimet, amely szabályozza a kalcium beépülését a tojásokba, így a madarak tojásai vékonyfalúak lettek és nem bírták ki a költési időszakot. Általánosságban azt mondhatjuk, hogy ugyan a halogénezett származékokra kémiai és biológiai lebomlási és metabolizációs mechanizmus ismeretes, a poliklórszármazékok peszticidként való további alkalmazása lipofil tulajdonságuk, perzisztenciájuk és számos metabolizációs termékük még teljesen nem felderített biológiai hatása miatt erősen kérdésessé vált. Ezekre a vegyületekre jellemző, hogy a környezetben mutatott perzisztenciájuk a C-Cl kötések számának növekedésével egyre kifejezettebb.
A 2,4,5-triklórfenoxi-ecetsavat defóliáns szerként használták. Gyártáskor az 1,2,4,5-tetraklórbenzol lúgos hidrolízisével állították elő a megfelelő fenolt. A folyamat hőmérsékleti viszonyait azonban gondosan kontrollálni kell. A reakció 1976-ban Sevesóban (Olaszország) „megszaladt” és a gyárban robbanás történt. A levegőbe több tíz kilogramm rendkívül mérgező 2,3,7,8-tetraklórdibenzodioxin (TCDD) került. A mérgezés számos vadon élő és háziállat pusztulásához, a lakosság körében súlyos bőrelváltozásokhoz és daganatos megbetegedésekhez vezetett.
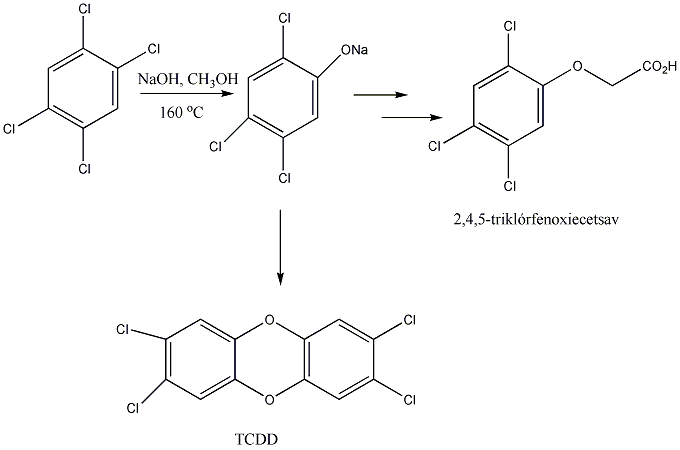
A redox, hidrolitikus ill. enzimkatalizált lebontási folyamatokat a legfontosabb, legnagyobb mennyiségben a természetbe kerülő antropogén szennyezőkön keresztül mutatjuk be. A következőkben egy-egy szénhidrogén, növényvédőszer és tenzid átalakulására mutatunk be néhány példát.
Az alifás és cikloalifás szénhidrogének (alkánok, alkének, alkinok, naftének) mint az ásványolaj alkotói a bányászat, a szállítás, a feldolgozás és a felhasználás komplex láncolatán haladnak keresztül. Az olajszennyezés káros hatásának lényege abban fogalmazható meg, hogyha vízzel érintkezik (pl. tankhajók katasztrófája) a víz felületén emulziós réteg képződik, amelyből szilárd és folyékony szénhidrogén-aggregátumok csapódnak ki. A víz-levegő határfelület hidrofóbbá válik, és az atmoszféra és a hidroszféra között a gázcsere korlátozódik („olajpestis”), a tengeri madarak és emlősök, valamint a halak életfeltételei drasztikusan romlanak. Az alkánok és a cikloalkánok biológiai lebomlásának sebességét a szénlánc szerkezete határozza meg. Elágazás nélküli, rövid, illetve közepes szénláncok mikroorganizmusok hatására gyorsan metabolizálódnak; az elbomlás leggyakrabban az egyik terminális metilcsoport oxidációjával kezdődik, s primer alkoholon, aldehiden keresztül karbonsavhoz vezet, amely azután -oxidáció révén továbbalakul. Ezzel szemben az elágazó szénláncú szénhidrogének perzisztenciája az elágazás fokától függően jelentősen megnövekedhet. Ezen utóbbiak jelenléte elsősorban az értékes, nagy oktánszámú motorhajtó anyagokra jellemző.
A viszonylag kis molekulatömegű aromás szénhidrogének (C6-C10-aromások: benzol, toluol, xilol, etil-benzol, sztirol, naftalin) a szerves kémiai technológia fontos közbülső-, illetve végtermékei. Azt feltételezik, hogy ezekből évente 1,5-2 Mt jut a tengervízbe. A kis molekulatömegű aromások – vízben való részleges oldhatóságuk következtében (100-1800 g m-3 20 ºC-on) – a tengervízzel nagy távolságokra is eljuthatnak. A felszíni vizekben és az ivóvízben eddig több mint száz, különböző benzolszármazékot azonosítottak. Az illékony aromások hatása elsősorban abban nyilvánul meg, hogy a víznek kellemetlen ízt és szagot kölcsönöznek. A mikroorganizmusok ezeket, a csak nagyobb koncentrációban fitotoxikus származékokat lebontják. Ezzel szemben ismeretes a benzolszármazékok sokirányú mérgező hatása az emlősökre és az emberre. A krónikus és akut intoxikáció számos esetét leírták, amelyek a központi idegrendszert károsító hatásra, karcinogén és mutagén elváltozásra, leukémiára, a szem irritálására és bőrmegbetegedésre utalnak.
Policiklusos aromás szénhidrogéneket (PAH) célirányosan csak igen kevés esetben állítanak elő, azonban mindig keletkeznek, ha szén- és hidrogéntartalmú kiindulási anyagokat dehidrogénező körülmények között >700 ºC hőmérsékletre hevítünk (pirolízis, tökéletlen égés, kokszosítás). Ilyenkor gázfázisban terjednek tova, a talajokra kiülepednek, és csekély vízoldhatóságuk ellenére (<5µg/1) a felszíni vizekben nagy területen szétoszlanak, mivel a kolloidális szemcsék adszorbeálják vagy felületaktív anyagok oldatba viszik őket. A kondenzált aromás vegyületek gyakran erős lipofil hatást mutatnak, s ennek következtében a szövetekben nagy bioakkumulációs faktorral feldúsulnak. A bioszférából ezek a származékok főként fotokémiai oxidáció vagy mikrobiológiai átalakulás révén távoznak.
A policiklusos aromás vegyületek – mindenekelőtt néhány képviselőjük bizonyított karcinogén hatása miatt – ökológiai szempontból kiemelt figyelmet érdemelnek. Ilyen vegyület pl. a benzol[a]pirén és az alábbi ábrán bemutatott vegyületek hatása jelentős, amelyeknek koncentrációja a felületi vízben ≤ 0,4 µg kg-1, vízi organizmusokban ≤ 100 µg kg-1, talajokban 3 µg kg-1 és erősen iparosodott területek vízi üledékeiben akár 3000 µg kg-1 is lehet. A benzol[a]pirén karcinogén aktivitása mechanizmusának magyarázatára azt feltételezik, hogy enzimkatalizált metabolizációja során epoxidáció, oxigéneződés, majd ismét epoxidáció révén trihidroxi-karbokation alakul ki, amely a DNS nukleofil komponenseivel (elsősorban guaninnal) vagy a RNS-val reagál.
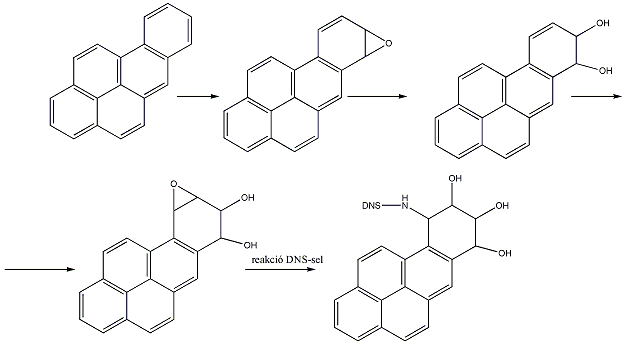
Az acetilkolinészteráz-gátlóként ismert foszforsav- és trifoszforsav-észterek pl. paration mikrobiológiai lebontása fontos folyamat, amelyben az oxidáció, redukció és hidrolízis meghatározó szerepet játszik. A paration átalakulásakor köztitermékként mindenekelőtt etanol, 4-nitro-, illetve 4-amino-fenol és szervetlen foszfát keletkezik. Az oxidatív deszulfurálódáskor keletkező köztitermék paraoxon (foszforsav-dietil-4-nitro-fenil-észter) mérgezőbb, mint a kiindulási vegyület.
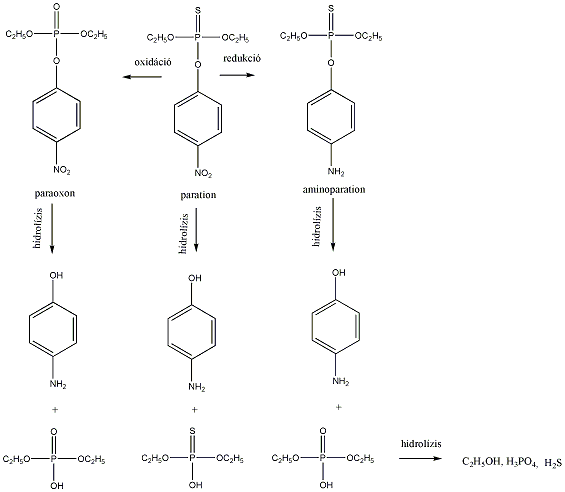
Napjainkban a mezőgazdaság nagy mennyiségben használ fel aromás nitrogénvegyület alapú herbicideket (szubsztituált fenil-karbamid, karbonsavanilid és N-fenil-karbaminsav-észter). Ezek lebontása során az amidázok közreműködésével elsőként a CO-NH kötés hidrolízise következik be. A primer reakciólépésben keletkező szubsztituált anilinek további sorsa attól függ, hogy mikrobiológiai támadással szemben mennyire ellenállók. Egyszerű anilinek pirokatechinné alakulnak át, majd ezt követően gyűrűfelhasadás következik be, végsősoron környezetbarát vízzé, széndioxiddá, ammóniává alakulnak. A nagy perzisztenciájú klórszubsztituált anilinek (3,4-diklóranilin) az aromás rendszert ellenállóvá teszik oxidációval szemben, és a további reakciók során stabil környezetkemikáliává – tetraklór-azobenzollá, humátkomplexekké – alakulnak át.
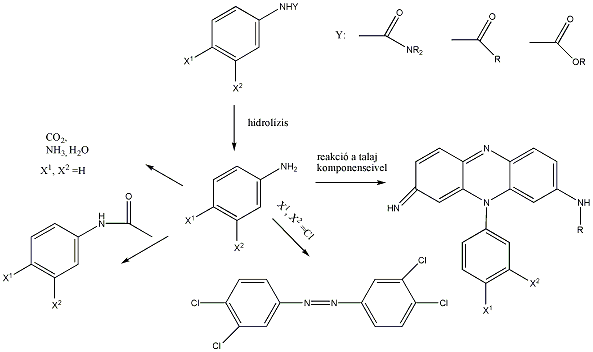
A tenzidek vízben részben jól oldódó felületaktív anyagok, amelyek a felületi feszültséget csökkentik, s ezen tulajdonságuk révén számos ipari folyamatban és a háztartásban (mosás, emulgeálás, diszpergálás, flotálás, hálóképzés stb.) alkalmazást nyernek. Szerkezetük alapvetően aszimmetrikus: hidrofil (poláris) fejcsoportból és hidrofób (apoláris) szénhidrogénláncokból állnak, amely utóbbi hosszúsága az egész molekula tulajdonságát befolyásolja. A poláris csoport milyensége szerint anionos, kationos, nemionos és amfoter detergenseket különböztetünk meg. A legnagyobb mennyiségben alkalmazott detergensek a következők: alkil-benzol-szulfonátok, alkán-szulfonátok, alkán–karboxilátok (zsírsavak nátriumsói), alkil-szulfátok, zsírsav-polietilén-glikol-észterek, zsíralkohol-polietilén-glikoléterek, alkilfenol-polietilén-glikoléterek és alkil-ammónium-sók.
A kereskedelmi mosó- és tisztítószerekben mintegy 1000 különféle anyag található. A tulajdonképpeni tenzid hatóanyag mellett komplexképzőket (builder: trifoszfát, zeolit, nitrilo-triacetát, etilén-diamin-tetraacetát), fehérítő adalékot (peroxo-borátok), korróziós inhibitort és stabilizátort (nátrium- és magnézium-szilikát), segédanyagot (optikai derítőket, lágyítókat), illatósító adalékokat, enzimeket és töltőanyagot (Na2SO4 stb.) tartalmaznak.
A tenzidek szennyvízzel való kijuttatása gyakorlatilag elkerülhetetlen, mivel tulajdonképpen az alkalmazás folyamán kvantitatíve nem használódnak el. A víz felületi feszültségének csökkentése és a habképződés számos élőlény számára végzetes. Éppen ezért érthető a mosószergyártók és –alkalmazók törekvése, hogy olyan detergenseket állítsanak elő, illetve alkalmazzanak, amelyek biológiailag gyorsan bomlanak. A felületaktív anyagként használatos alkil-benzol-szulfonátok,
R-C6H4-SO3 -Na+ a környezetben különböző perzisztenciát mutatnak mikrobiológiai lebontással szemben. Míg az elágazó láncú alkilszubsztituensek ezen az úton nehezen támadhatók, a lineáris származékot a mikroorganizmusok viszonylag gyorsan metabolizálják. A mineralizáció az oldallánc fokozatos feldarabolásával kezdődik, ω-, illetve β-oxidáció révén, majd az aromás gyűrű deszulfonálásával folytatódik, s végül szokásos mechanizmus szerint bekövetkezik a 4-es szénatom szubsztituált pirokatechin gyűrűfelhasadással járó degradációja.
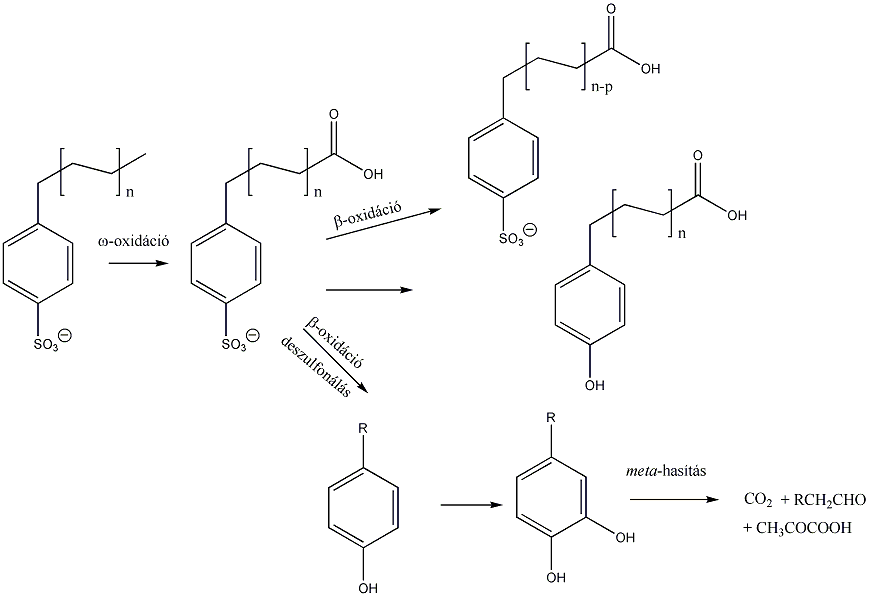
A szulfoncsoport leszakadása monooxigenáz-reakcióban játszódik le:
R-SO3-+O2+NADH + H+→ ROH + NAD++ HSO4 -.
Az alkil-benzol-szulfonátok mérgező hatása a halakra a szénlánc növekedésével erősen csökken. A három fő kritérium – mérgező hatás, tenzid hatás, biológiai degradáció – alapján a szénlánc hosszúságára optimumot határozhatunk meg. A szénatomok optimális száma 10-14 között van.
Mindezek alapján a természetben a következő enzimatikus oxidációs, redukciós és hidrolitikus folyamatokat az alábbiakban foglalhatjuk össze: